Keywords
RNA-seq, Android-based, simulations, mobile application, recommendations, experimental design,
RNA-seq, Android-based, simulations, mobile application, recommendations, experimental design,
RNA-seq offers several advantages over low-throughput technologies such as quantitative PCR and annotation-dependent methods such as microarrays. Designing RNA-seq experiments accurately, however, poses challenge to biologists. This is particularly true when prior knowledge on genome or transcriptome of the organism of choice is not available. It is important to determine the number of replicates and the number of sequencing reads, and choose the right analytical tool, to estimate subtle differences between expression levels of transcripts. In the current manuscript, we describe RNAtor, an Android app with a user-friendly graphical user interface (GUI) that helps biologists design RNA-seq experiments. RNAtor can be linked to any existing differential expression analysis tool, and can help design experiments to estimate expression differences of as low as 0.8–1.2X fold. RNAtor’s recommendations are based on an exhaustive combination of discovery with simulated reads for transcriptomes of varying sizes (3 to 100 MB), followed by validation with real datasets on wild-type and mutant conditions of Saccharomyces cerevisiae.
We simulated varying numbers of Illumina-like reads with replicates, with fold changes ranging from 1.2–5X between the control and treatment samples on a 3 MB human chr14 (hg19) transcriptome, using Polyester (Frazee et al., 2015). We detected differentially expressed genes (DEGs) on all the simulations using Tophat v2.1.1-Cufflinks v2.2.1 (Trapnell et al., 2012) based genome-guided workflow followed by differential expression analyses using five tools: Deseq v1.28.0 (Anders & Huber, 2010); Deseq2 v1.16.1 (Love et al., 2014); EdgeR v3.18.1 (Robinson et al., 2010); Cuffdiff-Cufflinks v2.2.1 (Trapnell et al., 2012); and Kallisto v0.43.1 (Bray et al., 2016) and a de novo assembly-based tool, Trinity v2.3.2 (Grabherr et al., 2011) followed by differential expression analyses using Kallisto v0.43.1 (Bray et al., 2016). We studied results from these simulations on the number of DEGs detected reliably and the extent of recovery of those DEGs. Based on these simulations, we arrived at recommendations on the number of reads, number of replicates, and the tool(s) needed to identify DEGs reliably. We validated these recommendations using simulated reads from larger transcriptomes (10MB, 30MB and 100MB), created by combining transcriptomes from more than one hg19 chromosome, and using a real Sacharomyces cerevisiae dataset (ENA accession: ERP004763) comprising of 48 biological replicates, for two conditions; wild-type (WT) and a snf2 knock-out (KO) mutant.
The size of the transcriptome (or genome if the transcriptome size is not known), taken from a user-defined or from a backend database, the number of replicates to use and the fold change of DEGs are user-defined parameters in RNAtor (Figure 1). An RNAtor flowchart highlighting simulation conditions and analytical tools used is provided in Supplementary Figure S1.
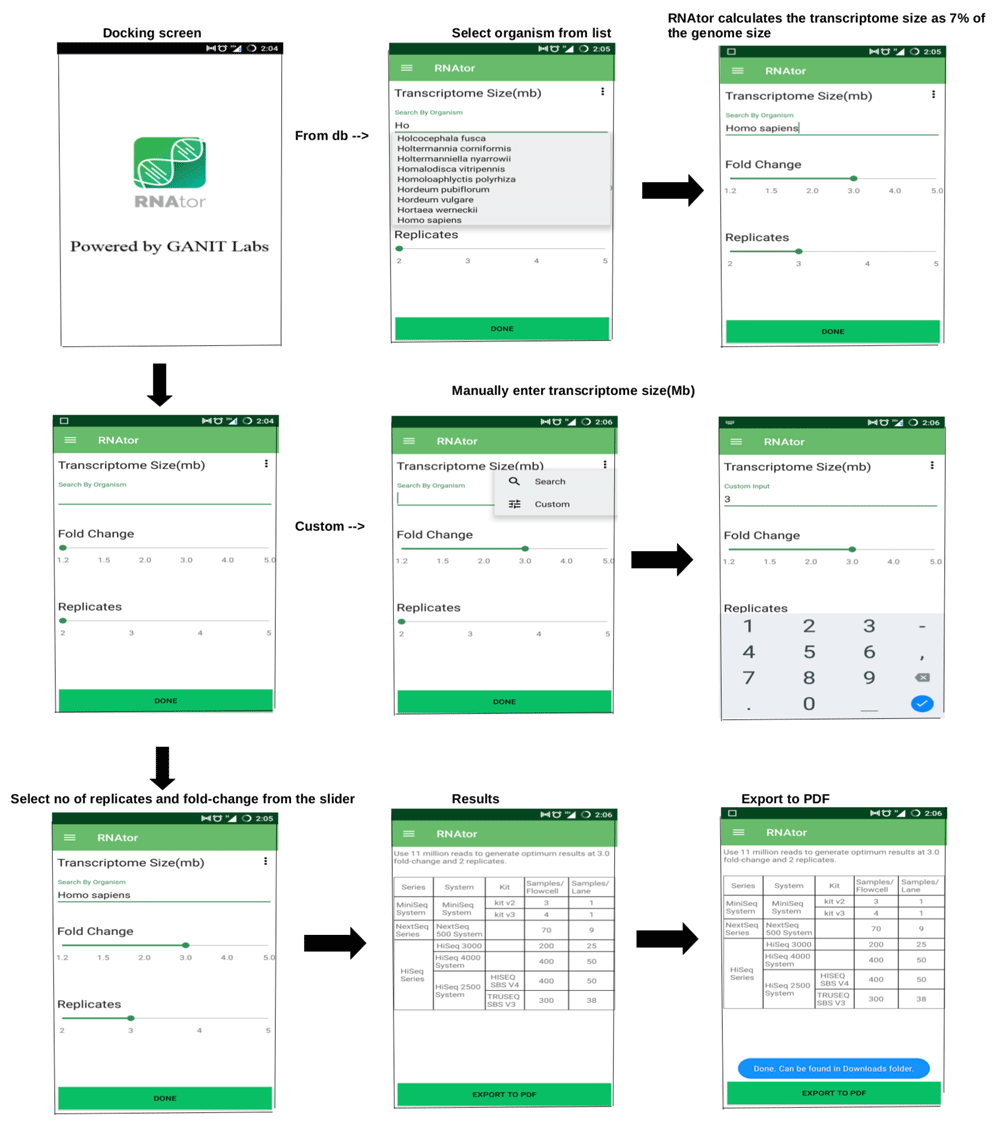
RNAtor was evaluated using questions that a biologist would typically ask before starting an experiment, followed by the recommendations provided by RNAtor.
1, 1.5, 6, 10, 14 and 20 million reads are needed for detection of differential expression of DEGs at 5-fold, 4-fold, 3-fold, 2-fold, 1.5-fold and 1.2-fold change, respectively, for a 3Mb transcriptome with 3 replicate samples.
We simulated 0.2–20 million reads for human chromosome 14 (~3Mb) and observed that the numbers of detected DEGs simulated at a given fold change peaked for a certain coverage before plateauing (Figure 2). This observation remained valid for the real data (Figure 3) and the large simulated transcriptomes (10Mb, 30Mb and 100Mb) (Supplementary Figure S2). Increasing the number of sequencing reads increased the sensitivity of detection. The final recommendations from RNAtor correspond to the number of DEGs at its peak, and are therefore a good compromise between sensitivity and keeping the cost of sequencing low. Changing the number of replicates does change the recommendation. For example, with more than three replicates, RNAtor suggests producing fewer reads to obtain the same information (Table 1).
| 2 replicates | 3 replicates | 4 replicates | 5 replicates | |
|---|---|---|---|---|
| 5fold | 6 | 2 | 1.5 | 1.5 |
| 4fold | 10 | 6 | 2 | 1.5 |
| 3fold | 10 | 6 | 6 | 6 |
| 2fold | 14 | 10 | 10 | 6 |
| 1.5fold | 30 | 20 | 20 | 14 |
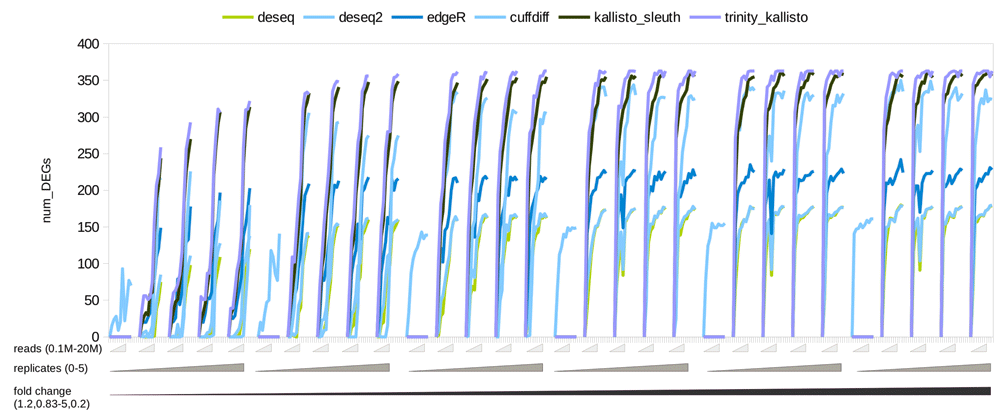
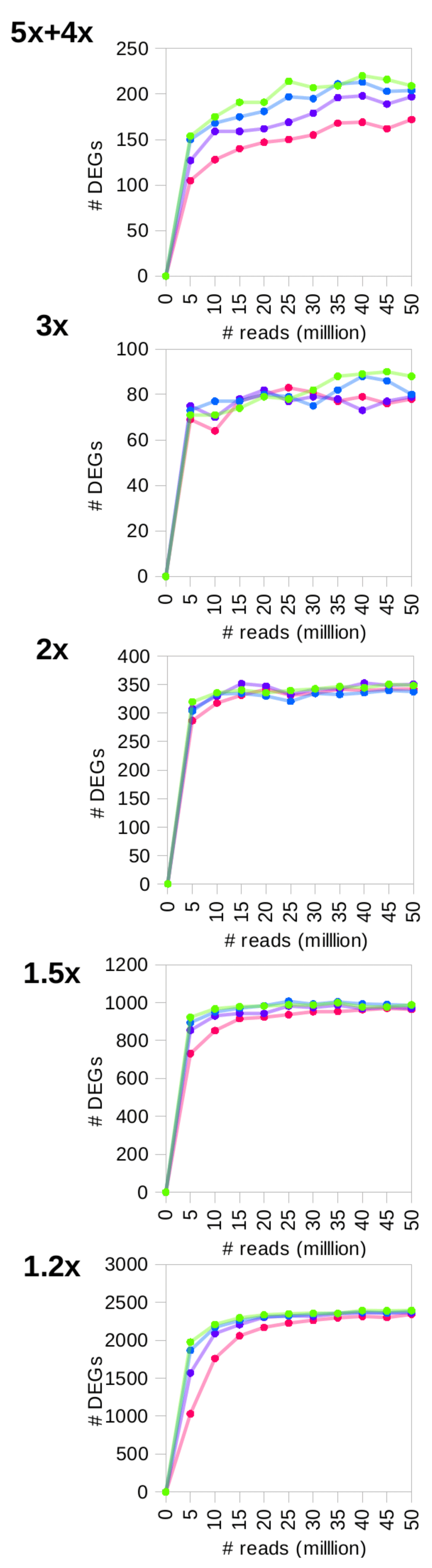
Kallisto detected optimal number of DEGs with the highest sensitivity. Focusing purely on the number of DEGs detected between WT and KO, Kallisto performed best over the other tools tested (Figure 2 and Supplementary Figure 3).
Cuffdiff can be used for high specificity and DeSeq2 and EdgeR, for high transcript recovery. Although Kallisto-Sleuth was fast and produced results with high sensitivity; we observed that this was at the expense of specificity of detection (Supplementary Figure S3). Cuffdiff produced results with high specificity albeit with a loss of sensitivity (Supplementary Figure S3). The transcript recovery was best for EdgeR among the 3 tools tested (CuffDiff, DeSeq and EdgeR, Supplementary Figure S4).
The assembly-based pipeline yields more DEGs with higher specificity. Using Trinity (Grabherr et al., 2011) as an assembly pipeline along with Kallisto enhanced the number of DEGs detected when compared with the genome-guided Kallisto-Sleuth pipeline (Figure 2). The specificity of Trinity-Kallisto was also better when compared to the Kallisto-Sleuth pipeline (Supplementary Figure S3).
Although some of the challenges with RNA-seq experiments have been addressed previously (Busby et al., 2013; Luo et al., 2014), currently there is no easy-to-use, biologist-friendly mobile phone-based app. Scotty, a previously reported, useful, interactive web-based tool aids RNA-seq experimental design. However, it has a dependence on pilot or prototype data, closely matching the actual experimental conditions (Busby et al., 2013). Additionally, it can detect genes or transcripts of only up to 2X fold change in the test condition relative to the control. RNAtor addresses some of these gaps as a user-friendly mobile app, however it has certain limitations. For example, it does not take into account the dynamic nature of any transcriptome (where the exact size of transcriptome is not known and cannot simply be derived from the genome size), the throughput of different sequencing instruments and the presence of spliced variants. These limitations will be addressed in future releases.
The Android version of RNAtor is available on Google Play Store.
Latest source code: https://github.com/binaypanda/RNAtor.
Archived source code as at the time of publication: https://doi.org/10.5281/zenodo.814905 (Panda, 2017).
License: RNAtor v1.0 is distributed under GNU GPLv3 licence.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)