Keywords
Fission yeast, H4K20me2, histone, methylation, DNA damage, checkpoint, telomere
Fission yeast, H4K20me2, histone, methylation, DNA damage, checkpoint, telomere
This version was revised to include data that rebut the objections of Ferreira and Nakamura, the two senior authors of the Carneiro et al. 2010 paper that claimed that H4K20me2 is excluded from Schizosaccharomyces pombe telomeres and we show is not the case. Due to an editorial error the link to dataset 2 in the version 1 article was not made available. Thus, we include the original Dataset 2 again in this version 2, for readers to see. Additionally, we provide Dataset 3 which contains the original dataset 2 along with two new spreadsheets which show that normalizing H4 ChIP data to input chromatin produces an artifact of low H4 levels for telomeric chromatin fragments. We also add a Supplementary Figure 1 showing that the available lot of Abcam ab9052 anti-H4K20me2 antibody has issues that make it a poor choice for ChIP compared to the Gentex GT282 antibody that we characterized and used.
See the authors' detailed response to the review by Miguel Godinho Ferreira and Toru M. Nakamura
The link to dataset 2 in the version 1 paper was not provided due to an editorial error. Thus, we now include below the original dataset 2 for readers to see (labelled Dataset 2), alongside the revised dataset 2 provided for the version 2 article (labelled Dataset 3).
Genome instability is a potentially lethal event for a eukaryotic cell, and a mutational force for genetic diseases such as cancer. DNA double-strand breaks (DSBs) can drive genome instability and are sensed by the DNA damage checkpoint, a defined set of evolutionarily-conserved proteins that bind the DSB to signal a pause or arrest of the cell cycle1 and recruit proteins to repair the DNA lesion2,3. Telomeres, the physical ends of linear eukaryotic chromosomes, are specialized DSBs that suppress DNA damage checkpoint activation by an unknown mechanism(s), even though telomeres are bound by many of the DNA damage checkpoint proteins that signal cell cycle arrest4. Carneiro et al. (Nature 467: 228-232) addressed this question using Schizosaccharomyces pombe cells that lack Taz1, the protein that binds to double-stranded telomere repeats5. Telomeres in taz1∆ cells have single-stranded DNA regions that are bound by checkpoint and DNA repair proteins, but cells do not arrest5,6. Immunofluorescence co-localization results from Carneiro et al. indicated that the ortholog of the human DNA damage checkpoint protein 53BP1 (Crb2) found at DSBs was not recruited to telomeres5. Crb2 can bind to dimethylated lysine 20 of histone H4 (H4K20me2) in nucleosomes7. Carneiro et al. presented data that H4K20me2 was depleted near telomeres in wild type and taz1∆ cells, suggesting a mechanism for checkpoint suppression5. Efforts to pursue this exciting result by ourselves and others failed. We therefore carefully re-evaluated the presence of H4K20me2 at different chromosomal loci, and found that H4K20me2 is not depleted near telomeres, indicating that checkpoint suppression occurs by some other mechanism(s).
Wild type (yJRE20-1) and histone H4 lysine 20 mutant (yJRE21-1) strains were previously described8 and were constructed as follows: The 5’ flanking region, the H4 gene, and the 3’ flanking region of each histone H4 gene were separately cloned into a pFA vector 5’ of the selection marker (hhf1 into pFA6a arg3MX6; hhf2 into pFA6a his3MX6; hhf3 into pFA6a ura4MX6). Approximately 500 base pairs 3’ to the initial fragment was cloned 3’ of the selection marker to the appropriate vector. The inclusive distance between the last A for the histone H4 TAA stop codon and the first G in the Asc I site from the pFA6a marker was 441 bp from H4.1 to arg3+, 707 bp from H4.2 to his3+ and 464 bp from H4.3 to ura4+ (plasmid maps are included in Supplementary File 1). Each construct was verified by restriction enzyme digestion and DNA sequencing of the fragments. Site-directed mutagenesis was used to mutate lysine 20 to arginine (H4K20R) at each gene copy; for wild type strains, the site was left unmutated. The resulting mutant constructs were verified by enzyme digestion and capillary dye terminator dideoxyDNA sequencing at ACGT (ACGT, Inc., Germantown, MD) to confirm the codon change corresponding to H4K20 (examples of aligned sequences are available in Supplementary File 2). Linearized fragments containing the 5’ fragment, selectable marker, and 3’ fragment were separately transformed into FY1645 (hhf1, h+) or FY1646 (hhf2 and hhf3, h-)9. Confirmation of integration was done by restriction digestion and DNA sequencing of the PCR product of the H4 gene. The strains with hhf1 (h+) and hhf3 (h-) marked and/or mutated were crossed to generate a strain in which hhf2 is the only unmarked gene copy. The resulting h+ strain was then crossed with the hhf2 (h-) marked strain to generate a strain in which all three loci of the histone H4 gene contain a selectable marker and are either wild type or mutated to arginine at lysine 20. Confirmation via digestion and DNA sequencing was performed after each cross. The H4K20R strain has been previously shown to be sensitive to DNA damaging agents8. The strains and primers used during strain construction are available upon request.
The strains used are described in Table 1. For the control strains lacking H4K20 methylation created by transformation for these experiments, two (yJRE141) or three (JA008) independent transformants were independently assayed in parallel. Cells were grown at 32°C in 300 ml in EMMG + AHRULK (yJRE141-3 and yJRF141-6) or EMMG + AHRULK + G418 (All other cells. EMMG is described in Moreno et al.10 and AHRULK + G418 contains 225 mg/l adenine, histidine, arginine, uracil, leucine, lysine and 200 mg/l G418 sulfate). Mid-log cells (9–12 × 106/ml or 0.8-1.2 OD600) were cross-linked with 1% formaldehyde for 15 min at room temperature and washed twice with cold HBS buffer (50 mM HEPES-NaOH pH 7.5, 140 mM NaCl). Cell pellets were stored at -80°C. All subsequent steps were performed at 4°C. Cell pellets were resuspended in ChIP-lysis buffer11 and lysed using mechanical disruption by bead-beater (Bio Spec Mini-Beadbeater-16) with 0.5 mm glass beads (Biospec 11079105) using 4 cycles of 45 sec followed by 60 sec on ice. The lysate was sonicated for 10 cycles on maximum power (30 sec ON and 59 sec OFF) in a Diagenode Bioruptor XL with sample tubes soaked in an ice water bath. Solubilized chromatin protein (2–4 mg) was used for each ChIP while 5 µl was saved as Input. Antibodies (2 µg) against H4K20me2 (GeneTex GT282 [RRID: AB_2728656] Lot #41582) or total histone H4 (Abcam ab10158 [RRID: AB_296888] Lot #GR133660-1) were mixed with chromatin and incubated at 4°C while rocking for 4 h. Dynabeads Protein G (50 µl, Life Technologies, Cat. No. REF 10004D) was then added and rocked overnight at 4°C. Beads were washed with ChIP lysis buffer, ChIP lysis buffer with 500 mM NaCl, Wash buffer and TE buffer (10 mM Tris, 1 mM EDTA pH 7.5) successively11. Beads were then resuspended in 145 µl of TES (1X TE with 1% SDS). Supernatant (120 µl) was recovered and incubated in a Thermomixer at 65°C, 1000 rpm (rotation per min) overnight to reverse cross-linking. For Input samples, TES buffer (115 µl) was added and incubated in the Thermomixer with the ChIP samples. Samples were treated with RNase A (2 µl of 10mg/ml added to each sample)(Roche 10109142001) for 15 min at 37°C and Proteinase K (2 µl of 20mg/ml added to each sample)(Roche 03115879001) for 30 min at 55°C, and purified by QIAgen PCR purification column (Cat.No. 28106)14. All samples from the same assay were processed for ChIP assay at the same time.
| Name | Genotype | Source |
|---|---|---|
| yJRE20-1 |
h- H4.1::arg3+ H4.2::his3+ H4.3::ura4+ ade6-210 arg3∆-4 his3∆-1 leu1-32 ura4-D18 | This lab8, used for western as WT |
| yJRE21-1 |
h- H4.1-K20R::arg3+ H4.2-K20R::his3+ H4.3-K20R::ura4+ ade6-210 arg3∆-4 his3∆-1 leu1-32 ura4-D18 | This lab8, used for western as H4K20R |
| ySLS298 | h- set9::CYC-terminator-kanMX (set9+ strain) | Greeson et al.12, used for western as set9-kan-wt |
| ySLS252 | h- set9∆::CYC-terminator -kanMX (set9-deletion strain) | Greeson et al.12, used for western as set9∆ |
| yNTG41 | h- set9-F178Y::CYC-terminator -kanMX | Greeson et al.12, used for western as set9-F178Y |
| yNTG39 | h- set9-F164Y::CYC-terminator -kanMX | Greeson et al.12, used for western as set9-F164Y |
| yNTG43 | h- set9-F195Y::CYC-terminator -kanMX | Greeson et al.12, used for western as set9-F195Y |
| yJRE210-1 |
h+ ade6-210 arg3-D4 his3-D1 leu1-32::pFA-LEU2-I-SceI ura4-D18 gal1-3':: ura4+-48bp TeloRpt-I-SceI-hph+ | This lab13, used for ChIP as wild type |
| JA002-3 | taz1∆::kanMX introduced into yJRE210-1 by transformation | This work, used for ChIP as taz1∆ |
| JA008-1 | set9∆::kanMX introduced into yJRE210-1 by transformation | This work, used for ChIP as set9∆ |
| JA008-2 | set9∆::kanMX in yJRE210-1, independent transformant from JA008-1 | This work, used for ChIP as set9∆ |
| JA008-3 |
set9∆::kanMX in yJRE210-1, independent transformant from JA008-1 and JA008-2 | This work, used for ChIP as set9∆ |
| yJRE141-3 |
h- ade6-210 arg3-D4 his3-D1 leu1-32 ura4-D18 hhf1K20R::arg3+ hhf2K20R:: his3+ hhf3K20R::natR leu1-32::pFA-LEU2-TETp-I-SceI gal1-3’::ura4+-48 bp TeloRpt-I-SceI-hph+ | This lab8, used for ChIP as H4K20R |
| yJRE141-6 | Independent isolate of yJRE141-3 | This lab8, used for ChIP as H4K20R |
Input samples were diluted to 1/100 with ddH2O while beads-only-ChIP, H4-ChIP and H4K20me2-ChIP samples were diluted to 1/10. Template DNAs (4 µl) were added to 5 µl of Roche LightCycler 480 SYBR Green I Master (2X) and primers (final concentration 0.6 µM) for a 10 µl total reaction volume. Each sample was run in triplicate on the same 384-well PCR plate (Roche LightCycler 480 Multiwell Plate 384, clear) in a Roche LightCycler 480. H4K20me2 immunoprecipitate levels were normalized to the total H4 immunoprecipitate levels at each locus15–17. The primers used are shown in Table 2, all primers were custom syntheses purchased from Integrated DNA Technologies (Skokie, IL, USA). Each locus was assayed using two or three primer pairs in the same qPCR assay for each ChIP, and the results for all primer pairs for a specific locus were averaged to obtain the final ChIP signal. The level of H4K20me2 at each locus was calculated as a ratio of H4K20me2 ChIP level to H4 ChIP level, where each ChIP level is expressed as a percent of input chromatin in the immunoprecipitated DNA (i.e. amount of DNA in H4K20me2 ChIP H4K20me2/amount DNA in the input chromatin divided by amount of DNA in H4 ChIP/amount DNA in the input chromatin).
All primers were custom syntheses purchased from IDTdna.com.
| Name | Sequence | Reference |
|---|---|---|
| 79 act1 1-1Fw | TGC CGA TCG TAT GCA AAA GG | Oya et al., 201318 |
| 80 act1-1Rev | CCG CTC TCA TAC TCT TG | Oya et al., 201318 |
| 139 act1-2Fw | GCA AGC GTG GTA TTT TGA CC | This study |
| 140 act1 2Rev | TCA GTC AAC AAG CAA GGG TG | This study |
| 141 act1-3Fw | TAC CAC TGG TAT CGT CTT GG | This study |
| 142 act1-3Rev | TAG TCA GTC AAG TCA CGA CC | This study |
| 143 hip3-1Fw | AGC CAA ATT TGA CGG TGT TC | This study |
| 144 hip3-1Rev | AGA CCT GGA CGG CAT TTT TA | This study |
| 145 hip3-2Fw | GGT GCC AAG ATT GTT TAT CCA | This study |
| 146 hip3-2Rev | ACG ACG TAT CCG ACA TCC TC | This study |
| 147 hip3-3Fw | ACG ATG CCG AGT AGT TCA GC | This study |
| 148 hip3-3Rev | TTC GTT GTT GTG TGC CTT TC | This study |
| 135 Telo-1Fw | CAG TAG TGC AGT GTA TTA TGA TAA AAA TGG | Carneiro et al., 20105 |
| 136 Telo-1Rev | CAG TAG TGC AGT GTA TTA TGA TAA TTA AAA TGG | Carneiro et al., 20105 |
| 121 Telo-2Fw | TAT TTC TTT ATT CAA CTT ACC GCA CTT C | Kanoh et al., 200519 |
| 122 Telo-2Rev | CAG TAG TGC AGT GTA TTA TGA TAA TTA AAA TGG | Kanoh et al., 200519 |
Cells of 5 OD (5 × 107 cells) were collected and resuspended in 200 µl SDS loading buffer without dye and reducing agent (50 mM Tris, 2% SDS, 10% glycerol). Cells were lysed using mechanical disruption by FastPrep 120 (Thermo Savant) with 0.5 mm glass beads, in cold room, using 2 cycles of 40 sec of disruption followed by 60 sec on ice. Cell lysis efficiency, monitored under microscope, always reached a minimum of 90%. The lysate was collected by poking holes on the bottom of the tubes and spinning into new tubes at 1000 rpm for 1 min at 4°C. The protein concentration was measured by BCA assay (Pierce 23225) on 96-well plate. After adding 4X SDS loading buffer, lysate of 10 µg was heated at 95°C for 5 min, spun down, and loaded into each lane on SDS-PAGE gel. The rest of the lysates were kept at -20°C.
Recombinant histone H4 (MLA-modified) H4K20me1 or H4K20me2 or H4K20me3 (Active Motif® 31224, 31225, 31226) and unmodified recombinant histone H4 (Active Motif® 31223) were resuspended in PBS buffer (in HPLC grade water) and used at the working concentration of 50 ng/μl except for H4K20me3 which was at 2.5 ng/μl. After adding 4X SDS loading buffer, 200 ng of recombinant histone H4 was heated at 95°C for 5 min, spun down, and loaded into each lane on SDS-PAGE gel. The rest of the proteins were stored at -20°C.
SDS-PAGE gels were prepared with a 15% resolving gel and a 4% stacking gel using 40% Acrylamide/Bis solution (BioRad 161-0146), Tris buffer and SDS. The gel was run in 1x SDS-Glycine-Tris running buffer with Odyssey One-Color Molecular Weight Protein Marker (Li-Cor 928-40000). The proteins were transferred onto nitrocellulose membrane (Li-Cor 926-31092) using Genie transfer system for 1 h with 1X transfer buffer with 20% methanol and 0.05% SDS. The membrane was stained with Ponceau S and the blot above the 25 kDa marker band was removed. The cut membrane was then rocked with Odyssey blocking buffer (Li-Cor 927-40000) at room temperature for 1 h, followed by incubation with anti-H4K20me2 antibodies (GeneTex GT282 [RRID: AB_2728656] Lot #41582) diluted 1:2000 in Odyssey blocking buffer with 0.2% Tween-20 at 4°C overnight. In some experiments, GT282 was replaced with Abcam ab9052 ([RRID:AB_1951942] lot #GR99672-1). Anti-H4 antibody (Abcam ab10158 [RRID: AB_296888] Lot #GR133660-1) was diluted at 1:10000 in Odyssey blocking buffer with 0.2% Tween-20. The anti-H4K20me2 blot was treated with the secondary antibody 680RD Goat anti-Mouse IgG (Li-Cor 926-68070 [RRID: AB_10956588]) in Odyssey blocking buffer with 0.2% Tween-20 at room temperature and rocked for 1 h and kept away from light during the incubation. For anti-H4 blots, the secondary antibody was Goat anti-Rabbit antibody IgG (800CW Li-Cor 926-32211 [RRID: AB_621843]). The blots were scanned by the Odyssey® CLx Imaging system to acquire Western blot signal and analyzed with the Image StudioTM software (v. 4.0).
If the DNA damage checkpoint at telomeres is severed by excluding H4K20me2 from telomeric chromatin, the presence of H4K20me2 in telomeric nucleosomes would activate the DNA damage response in taz1∆ cells, causing slower growth or cell cycle arrest. H4K20me2 constitutes ~25% of total H4 in S. pombe20, implicating a telomere-associated demethylase to deplete H4K20me2 at telomeres in taz1∆ cells to prevent checkpoint-mediated arrest. However, both a genome-wide screen of gene deletion mutants (D. Durocher, pers. comm.) and our screen of demethylase mutants failed to identify a mutant that caused taz1∆ cells to arrest, to grow poorly or to recruit more Crb2. We therefore re-evaluated H4K20me2 levels by chromatin immunoprecipitation (ChIP). We first identified commercial antibodies specific for H4K20me2 by western analysis using 11 different samples. Positive controls were extracts from wild type cells, cells where the single S. pombe H4K20 methylase gene set9 is marked and functional (set9-kan-wt) and recombinant H4 where the only modification is a chemical mimetic for K20me221. Negative controls included recombinant H4 where the only modifications were mimetics of 0, 1 or 3 methyl groups on lysine 20, and extracts from cells where all three copies of the H4 gene have lysine 20 mutated to arginine (H4K20R)8. A series of set9 mutants that methylate H4K20 to contain 0 (set9∆), 1 (set9-F164Y, set9-F178Y), or 1 and 2 methyl groups (set9-F195Y) were also assayed12. The specific antibody identified (Figure 1A) was used in ChIP to monitor H4K20me2 at the telomeric loci assayed in Carneiro et al. and two internal chromosomal loci in wild type and taz1∆ cells, and in mutants that lack H4K20 methylation, set9∆ and H4K20R. We also tested the antibody Carneiro et al. reported using to detect H4K20me2, Abcam ab9052. We found that this lot of ab9052 did not recapitulate the reactivity of the original antibody reported by Greeson12 for different set9 mutants and showed reduced reactivity to H4K20me2 (Supplementary Figure 1) compared to the GT282 antibody (Figure 1A), so this antibody was not used. Total H4 levels at these loci were monitored with an antibody that recognizes all H4 forms.
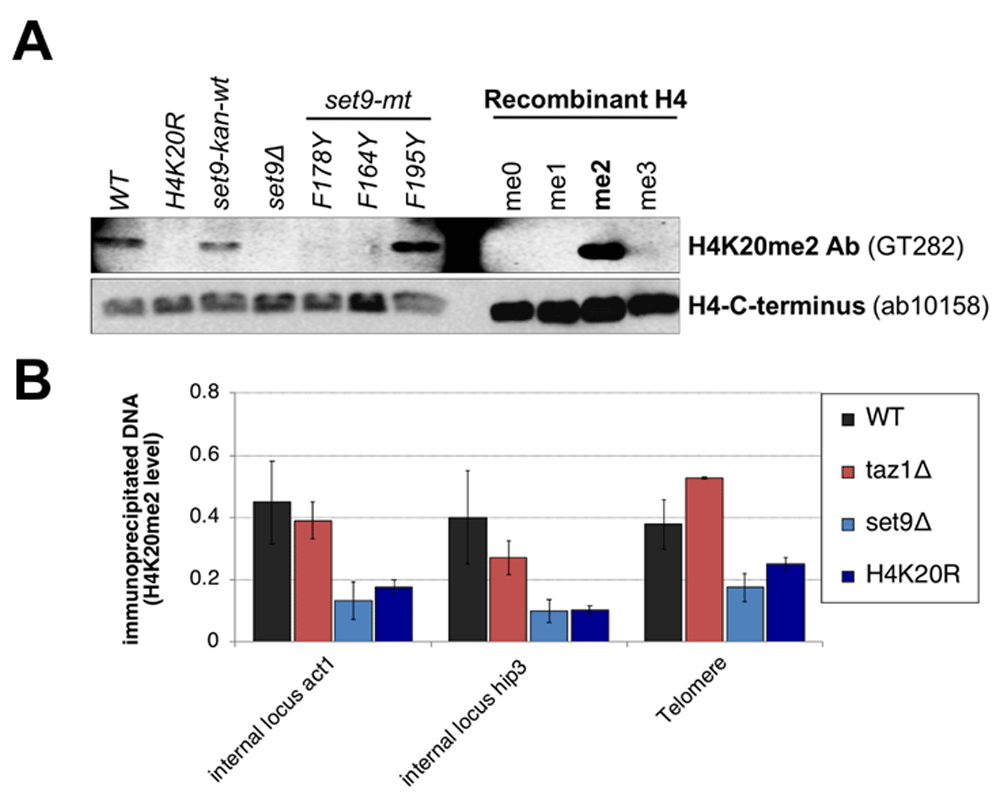
An antibody that specifically recognizes H4K20me2 was identified (A) and used in ChIP to measure levels of H4K20me2 in chromatin at two standard internal loci and loci adjacent to the telomere repeats (B). H4K20me2 levels are expressed as the ratio of the H4K20me2 ChIP levels (% of DNA in anti-H4K20me2 IP compared to input chromatin) over H4 ChIP levels (% of DNA in anti-histone H4 IP compared to input chromatin). Wild type and taz1∆ cells have the same levels at all loci and are clearly distinguishable from the negative controls (P values compared to wild type levels: all taz1∆ strains >0.18; all set9∆ and H4K20R strains ≤0.023, individual values are presented in Table 3). Each western blot in panel A used a separate, identically run gel to analyze the samples shown. M stands for molecular weight markers.
Assays were performed as described in Materials and Methods with two or more independent ChIP experiments. Each ChIP experiment was analyzed in triplicate. P values are from t-tests comparing each locus in a mutant strain to the same locus in the wild type strain, where values <0.05 are considered significant.
We found that H4K20me2 levels are similar at telomeres and the two internal loci in wild type and taz1∆ cells, and clearly distinguishable from the set9∆ and H4K20R negative controls (Figure 1B). These results were obtained by normalizing H4K20me2 signals to total H4 signals, which allows the direct comparison of this H4 modification at loci which contain nucleosomes. Carneiro et al. normalized to their ChIP signals to total input chromatin5. However, because telomere repeats are bound by non-nucleosomal proteins22, this normalization gives a much lower ChIP signal for total H4 and, thus, a lower signal for all H4 modifications. An example of this lower H4 signal is shown in the third spreadsheet of Dataset 3, where the level of H4 at wild type telomeres is 1/5 to 1/9 that of internal loci. Therefore, normalization of H4K20me2 ChIP signals to total H4 is necessary to monitor the fraction of modified H4 at telomeres.
The results in Figure 1B argue that while the damaged telomeres in taz1∆ cells block checkpoint activation, the mechanism is unlikely to be the suppression of H4K20me2 in telomeric chromatin. These results and conclusion are consistent with the genetic screen results that did not identify a demethylase required to sever the checkpoint in taz1∆ cells and suggest that searches for combinations of demethylase mutants that activate the checkpoint in taz1∆ cells will not be fruitful. Rather, broader approaches to investigate the differences between telomeres and DSBs may be required, including much more extensive characterization of the post-translation modifications of proteins at or near telomeres. While H4K20me2 levels are not reduced at telomeres, it is worth noting that checkpoint activation is the sum of multiple protein interactions and modifications, e.g. phosphorylation of histone H2A and modification of several checkpoint proteins23,24. Reducing the efficiency of some of these interactions may be sufficient to impair checkpoint signaling at taz1∆ cell telomeres. Results from such studies may provide an understanding of the anti-checkpoint activity of telomeres so that it may be modulated to treat telomere-related diseases such as cellular aging and cancer25.
A previous version of this article is available from bioRxiv - https://doi.org/10.1101/25138926
Dataset 1: unedited blot images 10.5256/f1000research.15166.d20937427
Dataset 2: Original excel workbook containing the Ct values from the PCRs and the location of the primers within the genes and telomere repeat adjacent DNA. 10.5256/f1000research.15166.d22080228
Dataset 3: Revised excel workbook containing the Ct values from the PCRs, the location of the primers within the genes and telomere repeat adjacent DNA and the ChIP values of total H4 normalized to input chromatin. 10.5256/f1000research.15166.d21973629
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)