Keywords
Biological Networks, Multiplex, Multi-layer, Community identification, Clustering, DREAM challenge
Biological Networks, Multiplex, Multi-layer, Community identification, Clustering, DREAM challenge
Biological macromolecules do not act isolated in cells, but interact with each other to perform their functions, in signaling or metabolic pathways, molecular complexes, or, more generally, biological processes. Thanks to the development of experimental techniques and to the extraction of knowledge accumulated in the literature, biological networks are nowadays assembled on a large-scale. A common feature of biological networks is their modularity, i.e., their organization around communities - or functional modules - of tightly connected genes/proteins implicated in the same biological processes1,2.
The Disease Module Identification (DMI) DREAM challenge aims at investigating different algorithms dedicated to the identification of communities, in a biomedical context3. The challenge has been divided into two sub-challenges, to identify communities either i) from six biological networks independently, or ii) from all these networks jointly. The clustering approaches proposed by the participants are assessed regarding their capacity to reveal disease communities, defined as communities significantly associated with genes implicated in diseases in GWAS studies3,4. The challengers proposed various strategies and clustering approaches, including kernel clustering, random walks or modularity optimization. We competed with an enhanced version of MolTi, a modularity-based software that we recently developed5. MolTi was initially developed to cluster multiplex networks, i.e., networks composed of different layers of interactions. It extended the modularity measure to multiplex networks and adapted the Louvain algorithm to optimize this multiplex-modularity. We have demonstrated that this multiplex approach better identifies the communities than approaches merging the networks, or performing consensus clusterings, both on simulated and real biological datasets5.
Grounded on these initial results, we here extended and tested our MolTi software, both on simulated data and on the DMI challenge framework. We improved MolTi with the implementation of a randomization procedure, the consideration of edge and layer weights, and a recursive clustering of the classes larger than a given size.
With simulated data, we observed that considering more than one network layer improves the detection of communities, as already noted5, but also that communities are better detected with the randomization procedure. With the DMI benchmark, we pointed to a great dependence on the GWAS dataset used for the evaluation and on the FDR threshold defined, but, overall, randomizations and edge and layer weights increase the number of detected disease communities.
We detected communities with an extended version of MolTi5, a modularity-based software. Although MolTi was specifically designed for multiplex networks, it deals with monoplex networks by considering them as multiplexes composed of a single layer. All the networks are here considered undirected. The new version of MolTi, MolTi-DREAM, and the scripts used for the DMI DREAM challenge are available at https://github.com/gilles-didier/MolTi-DREAM.
Modularity. Network modularity was initially designed to measure the quality of a partition into communities6, and subsequently used to find such communities. Since finding the partition optimizing the modularity is NP-complete, we applied the meta-heuristic Louvain algorithm7. Louvain starts from the community structure that separates all vertices. Next, it tries to move each vertex from its community to another, picks the move that increases the most modularity, and iterates until no change increases the modularity anymore. It then replaces the vertices by the detected communities and performs the same operations on the newly obtained graph, until the modularity cannot be increased anymore. In order to handle multiplexes, we use a multiplex-adapted modularity and an adaptation of the Louvain algorithm for optimizing this multiplex-modularity.
Edge and layer weights Modularity approaches can deal with weighted networks8, and we modified MolTi to handle weighted networks. We also added the possibility to weight each layer of the multiplex network: the contribution of each layer in Equation (1) is multiplied by its weight when computing the multiplex modularity.
Multiplex modularity The modularity measure to detect communities in a multiplex network (X(g))g can be written as
where X(g) denotes the (monoplex) network of the layer g, w(g) is the user-defined weight associated to the network g, m(g) is the sum of the weights of all the edges of X(g), is the weight of the edge {i, j} in X(g), is the sum of the weights of all the edges involving vertex i in X(g), δci,cj is equal to 1 if i and j belong to a same community and to 0 otherwise.
Randomization. We implemented a randomized version of the Louvain algorithm, similar to the one in GenLouvain9. Rather than updating the current partition by picking the move leading to the greatest increase of the modularity, we randomly pick a move among those leading to an increase of the modularity. Different runs of the randomized Louvain generally return different partitions, even if the results are often close. MolTi-DREAM runs the randomized Louvain algorithm a user-defined number of times (from one to ten in this work, four by default), and returns the partition with the highest modularity.
We simulated random multiplex networks with a fixed known community structure, namely 1,000 vertices split into 20 balanced communities, and various topological properties (i.e., dense/sparse/mixed, with/without missing data)5. Multiplex networks are simulated by drawing each layer according to this structure and fixed intra/inter community edge probabilities (0.1/0.01 for sparse layers and 0.5/0.2 for dense ones). We also generated multiplex networks with missing data in which we randomly withdrawn vertices of each layer with probability 0.5. The relevance of a community structure is assessed by computing the adjusted Rand index10 between the detected communities and the ones used to simulate the multiplex networks.
Biological Networks. The DMI challenge provided six biological networks: two protein-protein interactions, one signaling, one co-expression, one network linking genes essential for the same cancer types, and one network connecting evolutionary-related genes. These six networks have various sizes and edge densities (Table 1). All networks have weighted edges, and all networks but the signaling network are undirected. However, we considered the signaling network as undirected.
Evaluations with GWAS data. The communities identified by the different challengers were evaluated according to the associations of their member genes with GWAS data, following the PASCAL tool4. The procedure leverages the SNP-based p-value statistics obtained from 180 GWAS datasets, covering common diseases and traits. It is to note that an important parameter is the FDR threshold used to define the significant associations3,4, and to get the number of significant disease communities. We used three datasets: the “Leaderboard” (76 GWASs) and “Final” (104 GWASs), which were used during the challenge, and their union in a “Total” dataset (180 GWASs).
Obtaining modules in a given size range. The DMI challenge set up two constraints on the submitted communities: no overlap and a size ranging from 3 to 100 nodes. We tested different pre-filters (pruning leaves), parameters (resolution parameter, recursions, combination of graph weights for multiplexes) and post-filters (density, size, pruning leaves) in each leaderboard round. We took into account both the number of significant communities and the total number of submitted communities to evaluate the pertinence of each combination. All partitions were post-filtered to keep only classes containing from 7 to 100 nodes.
Resolution parameter Modularity-based clustering approaches are often associated to a resolution parameter γ to tune the size of the obtained communities. We tested different values of this parameters (γ = 1, γ = 5, γ = 10, γ = 110), but the leaderboard tests showed clearly better results for the recursive approach. We chose to keep the default γ = 1 and focused on this recursive procedure.
Recursion procedure We re-clustered all the communities above a certain size (here 100 vertices) by extracting the corresponding subgraphs from the networks and applying recursively the MolTi algorithm. We iterated the process until obtaining only communities with less than 100 vertices, if possible (some communities with more than 100 vertices cannot be split by considering modularity).
To evaluate the accuracy of the community structures detected from the initial MolTi and its improved version that includes the randomization procedure, we simulated random multiplex networks with a fixed, known community structure, and various features5. Considering a greater number of layers always improves the inference of communities, as already observed5 (Figure 1). In addition, communities are better detected from sparse multiplexes than from dense ones. We also observed that the randomizations improve the accuracy of the detected communities, in particular for dense multiplex networks, with or without missing data. Increasing the number of randomization runs improves the results, but to a limited extend after more than four runs.
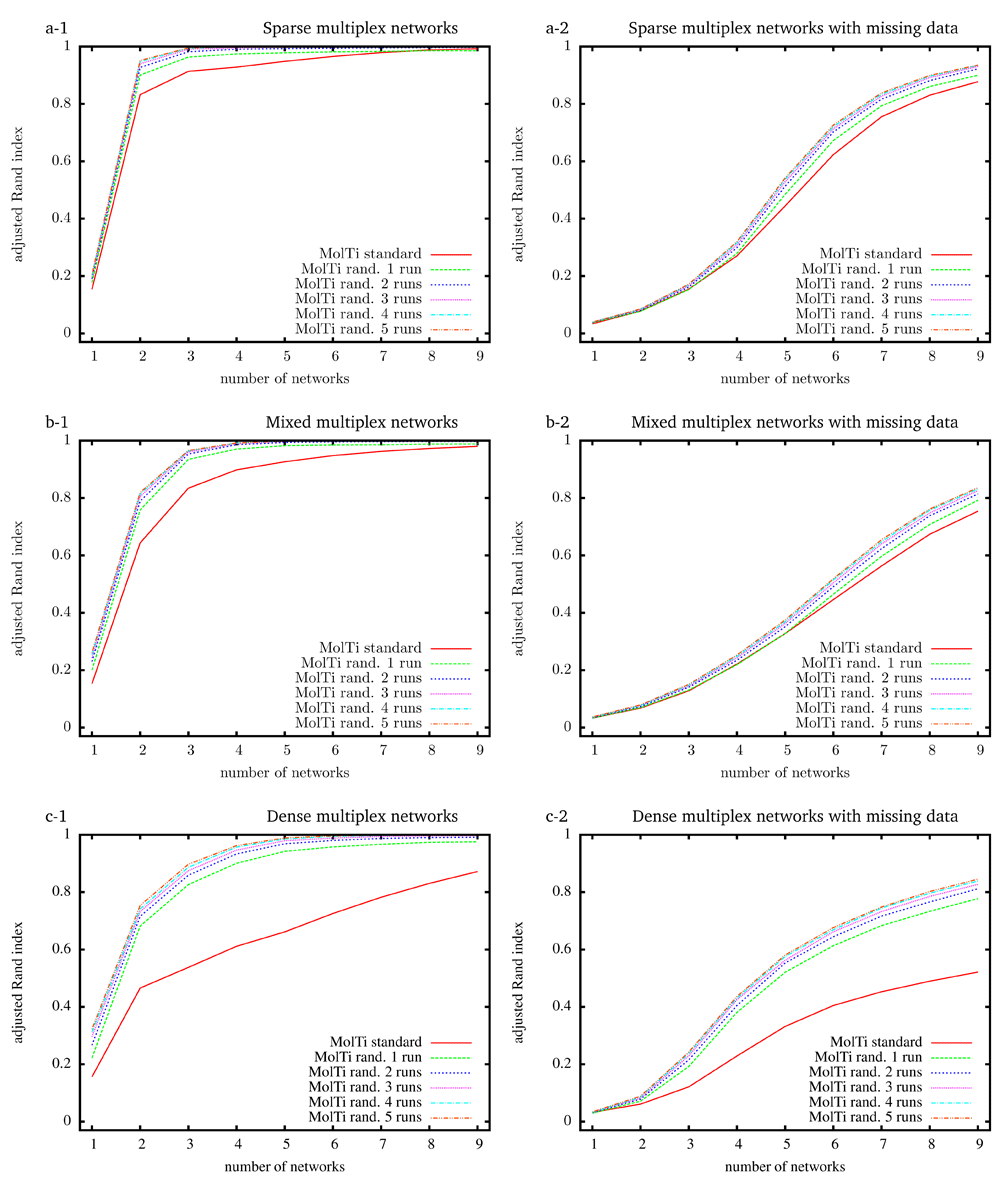
Multiplex networks contain from 1 to 9 graph layers. The indexes are averaged over 2,000 random multiplex networks of 1,000 vertices and 20 balanced communities. Each layer of sparse (resp. dense) multiplex networks is simulated with 0.1/0.01 (resp. 0.5/0.2) internal/external edge probabilities. Mixed multiplex networks are simulated by uniformly sampling each layer among these two pairs of edge probabilities. Multiplex networks with missing data (right column) are generated by withdrawing vertices from each layer with probability 0.5.
We applied the improved MolTi to the networks provided by the DMI challenge (Methods). We focused on the sub-challenge 2, which was dedicated to the identification of communities from multiple networks. We considered the six DMI biological networks as layers of a multiplex network, and applied the recursion procedure to obtain communities in the required size range. The significant disease communities were selected regarding their enrichments in GWAS-associated genes (Methods). We observed first that the number of detected disease communities varies in a non-trivial way depending on the GWAS dataset and FDR threshold used (Figure 2). However, we can observe that the number of detected significant disease modules slightly increases after randomization, in particular when the FDR threshold is higher (Figure 2).
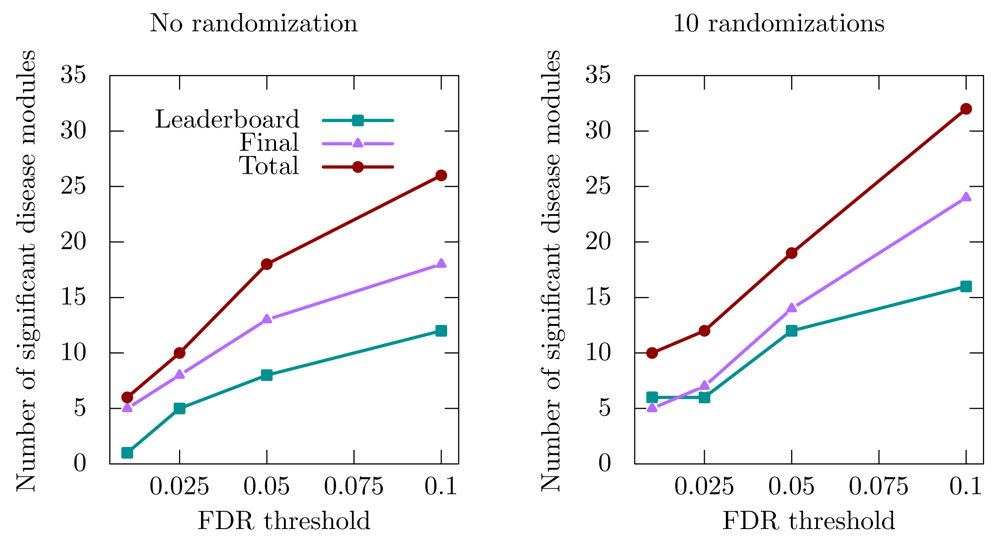
“Leaderboard” and “Final” datasets were used during the training and final evaluation of the challenge, respectively, whereas the “Total” dataset is the union of the two previous ones.
Multiplex versus monoplex. We next evaluated the added value of the multiplex approach as compared to the identification of modules from the individual networks. When analyzing the significant disease modules obtained for a FDR threshold of 0.1, we observed that combining biological networks in a multiplex generally increases the number of significant modules (Figure 3). However, this does not stand for the cancer and/or homology networks, which lower the number of significant modules retrieved when added as layers of the multiplex. We hypothesize that the community structures of these networks (if they exist) are so unrelated that it is pointless to seek for a common structure by integrating them.
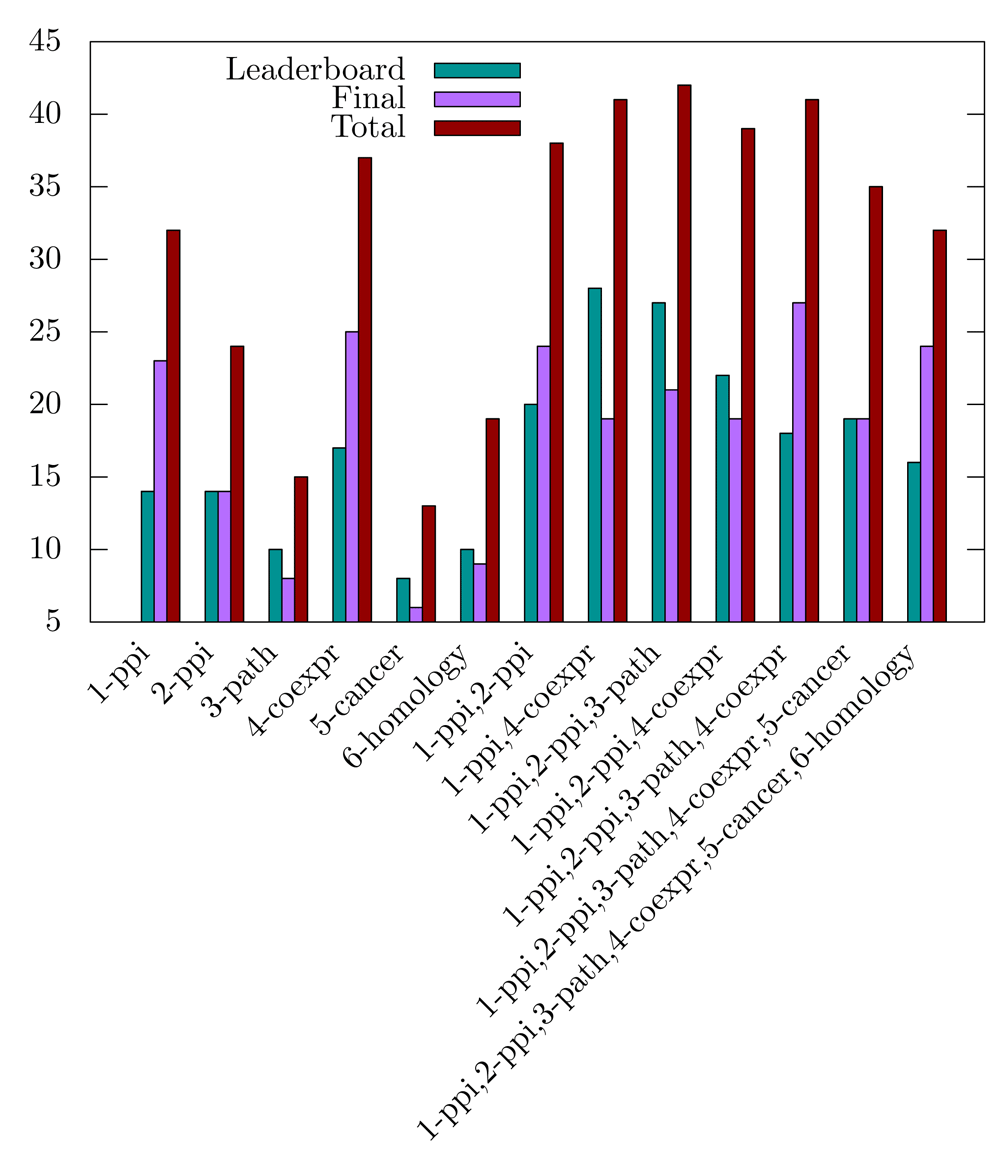
Ten randomization have been applied, and the FDR threshold is set to 0.1.
These observations are consistent with the DMI challenge observations, in which the top-scoring team in the sub-challenge 2 handled only the two protein-protein interaction networks. Our algorithm also performs well with the two protein-protein interactions networks, but the highest number of disease modules is retrieved by considering network combinations that exclude the cancer and homology network layers (Figure 3).
Evaluation of the edge and layer weighting. All the six biological networks used in the DMI challenge have weighted edges. We compared the number of disease modules obtained by considering or not these weights in the MolTi partitioning, for different FDR thresholds (Table 2). We observed that intra-layer edge weights only has a slight effect on the number of identified significant disease modules, except for the very low significance threshold of 0.01, where it seems pertinent to use these weights.
| FDR | Unweighted | Weighted |
|---|---|---|
| 0.01 | 5 | 10 |
| 0.025 | 13 | 12 |
| 0.05 | 20 | 19 |
| 0.1 | 30 | 32 |
MolTi-DREAM allows assigning weights to each layer of the multiplex network, for instance to emphasize the layers known to contain more relevant biological information. Given the results of the DMI challenge and our first analyses, we decided to test a combination of weights that would lower the importance of the 5-cancer and 6-homology network layers. We observed that this led to detecting more disease modules (Figure 4). Conversely, less disease modules are detected when higher weights are given to these networks (Figure 4).
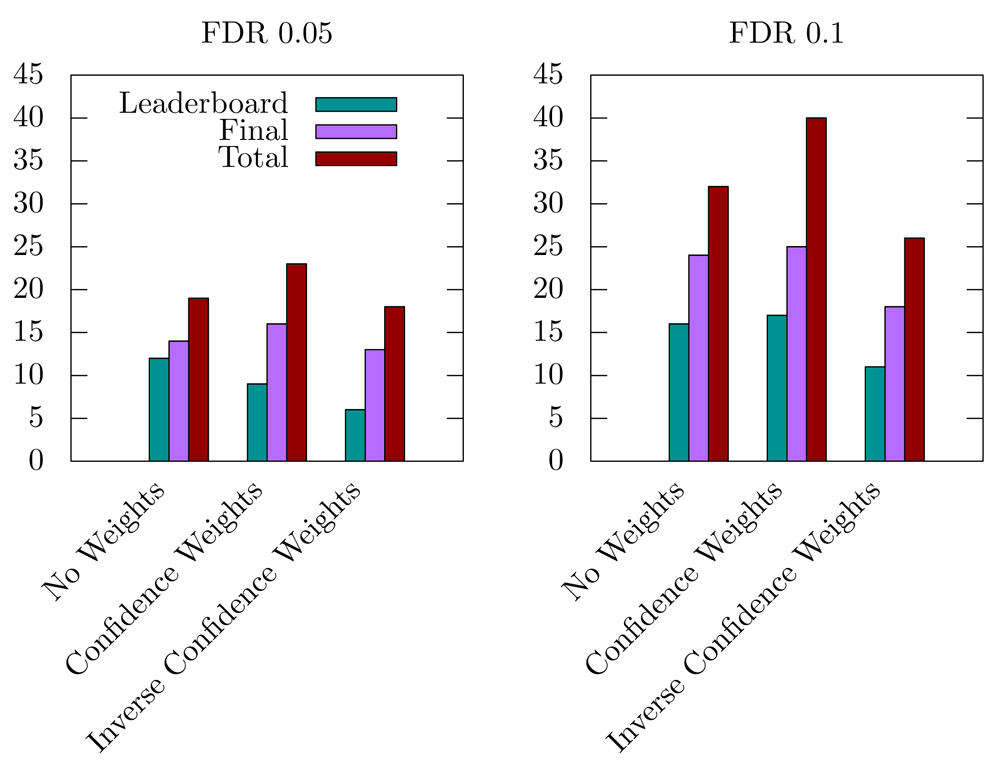
We applied here the MolTi software and various extensions to identify disease-associated communities following the DMI challenge benchmark. The new version of MolTi, MolTi-DREAM, runs a randomization procedure, takes into account edge and layer weights, and performs a recursive clustering of the classes that are larger than a given size. We finished tied for second in the challenge. However, even if we obtained higher scores than monoplex approaches, the difference was not significant and the organizers of the DREAM challenge declared the sub-challenge 2 vacant.
In the simulations, all the networks are randomly generated from the same community structure. These networks can thereby be seen as different and partial views of the same underlying community structure. Combining their information in a suitable way is thereby expected to recover the original structure more accurately. In contrast, combining networks with unrelated community structures (or no structure at all) is rather likely to blur the signal carried by each network. The DMI biological networks are constructed from different biological sources that might correspond to unrelated community structures.
This may explain the results of the sub-challenge 2, in which the top-performer used only the two protein-protein interaction networks, and the fact that the highest number of modules retrieved by our approach was not obtained from a multiplex containing all the six networks. From a biological perspective, the protein-protein networks and the pathway networks are expected to contain mainly physical or signaling interactions between proteins. It has been shown that interacting proteins tend to be co-expressed11, which could explain why the co-expression network also provides complementary information. In contrast, both the cancer and the homology networks are determined from processes operating at a very different level.
Evaluating the relevance of the community structure detected from real-life datasets is a very complicated problem since the actual structure is hidden and generally unknown. In this context, the only possibility for assessing the detected communities is to consider indirect evidence provided by some independent biological information. Different teams are thereby developing proxies to evaluate the communities, mainly based on testing the enrichment of genes contained in each community in Pathways or Gene Ontology annotations. The approach followed by the DMI DREAM challenge is based on GWAS data. This GWAS-based evaluation is specific in the sense that it considers p-value-weighted annotations rather than usual binary ones, i.e., “annotated/not annotated”. This probably contributed to the volatility of the results observed with the DMI DREAM challenge framework.
MolTi-DREAM and the scripts used for the DMI DREAM challenge: https://github.com/gilles-didier/MolTi-DREAM
Archived scripts and source code for MolTi-DREAM as at time of publication: http://doi.org/10.5281/zenodo.130120912
License for MolTi-DREAM: GNU 3
GD designed MolTi and its extensions, AB and AV applied MolTi during and after the challenge. AV and AB are currently at Aix*Marseille Univ, Inserm, MMG, France. All authors participated to the design of the study, the interpretation of the results and the writing of the manuscript.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)