Keywords
bioconductor, r, rstats, regulatory genomics, functional genomics, genetics, gwas, transcriptomics, integration, multiomics
This article is included in the Bioconductor gateway.
This article is included in the RPackage gateway.
bioconductor, r, rstats, regulatory genomics, functional genomics, genetics, gwas, transcriptomics, integration, multiomics
CAGE: cap analysis of gene expression; DHS: DNase I hypersensitive site; eQTL: expression quantitative trait locus; GWAS: genome-wide association study; PheWAS: phenome-wide association study; SLE: systemic lupus erythematosus; SNP: single nucleotide polymorphism; TSS: transcription start site
Discovering and bringing new drugs to the market is a long, expensive and inefficient process1,2. Increasing the success rates of drug discovery programmes would be transformative to the pharmaceutical industry and significantly improve patients’ access to medicines. Of note, the majority of drug discovery programmes fail for efficacy reasons3, with up to 40% of these failures due to lack of a clear link between the target and the disease under investigation4.
Target selection, the first step in drug discovery programmes, is thus a critical decision point. It has previously been shown that therapeutic targets with a genetic link to the disease under investigation are more likely to progress through the drug discovery pipeline, suggesting that genetics can be used as a tool to prioritise and validate drug targets in early discovery5,6.
Over the last decade, genome-wide association studies (GWASs) have revolutionised the field of human genetics, allowing to survey DNA mutations associated with disease and other complex traits on an unprecedented scale7. Similarly, phenome-wide association studies (PheWAS) are emerging as a complementary methodology to decipher the genetic bases of the human phenome8. While many of these associations might not actually be relevant for the disease aetiology9, these methods hold much promise to guide pharmaceutical scientists towards the next generation of drug targets10.
Arguably, one of the biggest challenges in translating findings from GWASs to therapies is that the great majority of single nucleotide polymorphisms (SNPs) associated with disease are found in non-coding regions of the genome, and therefore cannot be easily linked to a target gene11. Many of these SNPs could be regulatory variants, affecting the expression of nearby or distal genes by interfering with the process of transcription (e.g.: binding of transcription factors at promoters or enhancers)12.
The most established way to map disease-associated regulatory variants to target genes is probably to use expression quantitative trait loci (eQTLs)13, variants that affect the expression of specific genes. Over the last few years, the GTEx consortium assembled a valuable resource by performing large-scale mapping of genome-wide correlations between genetic variants and gene expression across 44 human tissues14.
However, depending on the power of the study, it might not be possible to detect all existing regulatory variants as eQTLs. An alternative is to use information on the location of promoters and distal enhancers across the genome and link these regulatory elements to their target genes. Large, multi-centre initiatives such as ENCODE15, Roadmap Epigenomics16 and BLUEPRINT17,18 mapped regulatory elements in the genome by profiling a number of chromatin features, including DNase hypersensitive sites (DHSs), several types of histone marks and binding of chromatin-associated proteins in a large number of cell lines, primary cell types and tissues. Similarly, the FANTOM consortium used cap analysis of gene expression (CAGE) to identify promoters and enhancers across hundreds of cells and tissues19.
Knowing that a certain stretch of DNA is an enhancer is however not informative of the target gene(s). One way to infer links between enhancers and promoters in silico is to identify significant correlations across a large panel of cell types, an approach that was used for distal and promoter DHSs20 as well as for CAGE-defined promoters and enhancers21. Experimental methods to assay interactions between regulatory elements also exist. Chromatin interaction analysis by paired-end tag sequencing (ChIA-PET)22,23 couples chromatin immunoprecipitation with DNA ligation and sequencing to identify regions of DNA that are interacting thanks to the binding of a specific protein. Promoter capture Hi-C24,25 extends chromatin conformation capture by using “baits” to enrich for promoter interactions and increase resolution.
Overall, linking genetic variants to their candidate target genes is not straightforward, not only because of the complexity of the human genome and transcriptional regulation, but also because of the variety of data types and approaches that can be used. To address this, we developed STOPGAP (systematic target opportunity assessment by genetic association predictions), a database of disease variants mapped to their most likely target gene(s) using different types of regulatory genomic data26. The database is currently undergoing a major overhaul and will eventually be superseded by POSTGAP. A similar resource and valid alternative is INFERNO (inferring the molecular mechanisms of noncoding variants)27.
In this workflow, we will explore how regulatory genomic data can be used to connect the genetic and transcriptional layers by providing a framework for the functional annotation of SNPs from GWASs. We will use eQTL data from GTEx14, FANTOM5 correlations between promoters and enhancers21 and promoter capture Hi-C data25.
We start with a common scenario: we ran a RNA-seq experiment comparing patients with a disease and healthy individuals, and would like to discover key disease genes and potential therapeutic targets by integrating genetic information in our analysis.
R version 3.4.2 and Bioconductor version 3.6 were used for the analysis. The code below will install all required packages and dependencies from Bioconductor and CRAN:
source("https://bioconductor.org/biocLite.R") # uncomment the following line to install packages #biocLite(c("DESeq2", "GenomicFeatures", "GenomicRanges", "ggplot2", "gwascat", "recount", "pheatmap", "RColorBrewer", "rtracklayer", "R.utils", "splitstackshape", "VariantAnnotation"))
The RNA-seq data we will be using comes from blood of patients with systemic lupus erythematosus (SLE) and healthy controls28.
We are going to use recount29 to obtain gene-level counts:
library(recount) # uncomment the following line to download dataset #download_study("SRP062966") load(file.path("SRP062966", "rse_gene.RData")) rse <- scale_counts(rse_gene) rse ## class: RangedSummarizedExperiment ## dim: 58037 117 ## metadata(0): ## assays(1): counts ## rownames(58037): ENSG00000000003.14 ENSG00000000005.5 ... ## ENSG00000283698.1 ENSG00000283699.1 ## rowData names(3): gene_id bp_length symbol ## colnames(117): SRR2443263 SRR2443262 ... SRR2443147 SRR2443149 ## colData names(21): project sample ... title characteristics
Other Bioconductor packages that can be used to access data from gene expression experiments directly in R are GEOquery30 and ArrayExpress31.
So, we have 117 samples. This is what the data looks like:
assay(rse)[1:10, 1:10] ## SRR2443263 SRR2443262 SRR2443261 SRR2443260 SRR2443259 ## ENSG00000000003.14 19 6 10 10 8 ## ENSG00000000005.5 0 0 0 0 0 ## ENSG00000000419.12 489 238 224 323 281 ## ENSG00000000457.13 594 503 530 670 775 ## ENSG00000000460.16 232 173 166 252 268 ## ENSG00000000938.12 21554 18918 14260 19869 26586 ## ENSG00000000971.15 94 57 45 59 35 ## ENSG00000001036.13 500 397 358 407 500 ## ENSG00000001084.10 373 298 336 367 391 ## ENSG00000001167.14 827 832 837 1091 1013 ## SRR2443258 SRR2443257 SRR2443256 SRR2443255 SRR2443254 ## ENSG00000000003.14 6 2 24 21 11 ## ENSG00000000005.5 0 0 0 0 0 ## ENSG00000000419.12 333 214 390 270 359 ## ENSG00000000457.13 712 461 603 613 609 ## ENSG00000000460.16 263 160 228 245 234 ## ENSG00000000938.12 17377 19981 15136 13039 16994 ## ENSG00000000971.15 76 26 53 60 50 ## ENSG00000001036.13 714 364 575 438 638 ## ENSG00000001084.10 535 326 581 438 418 ## ENSG00000001167.14 967 737 874 886 902
We note that genes are annotated using the GENCODE32 v25 annotation, which will be useful later on. Let’s look at the metadata to check how we can split samples between cases and controls:
colData(rse) ## DataFrame with 117 rows and 21 columns ## project sample experiment run ## <character> <character> <character> <character> ## SRR2443263 SRP062966 SRS1048033 SRX1168388 SRR2443263 ## SRR2443262 SRP062966 SRS1048034 SRX1168387 SRR2443262 ## SRR2443261 SRP062966 SRS1048035 SRX1168386 SRR2443261 ## SRR2443260 SRP062966 SRS1048036 SRX1168385 SRR2443260 ## SRR2443259 SRP062966 SRS1048037 SRX1168384 SRR2443259 ## ... ... ... ... ... ## SRR2443151 SRP062966 SRS1048145 SRX1168276 SRR2443151 ## SRR2443150 SRP062966 SRS1048146 SRX1168275 SRR2443150 ## SRR2443148 SRP062966 SRS1048147 SRX1168273 SRR2443148 ## SRR2443147 SRP062966 SRS1048148 SRX1168272 SRR2443147 ## SRR2443149 SRP062966 SRS1048149 SRX1168274 SRR2443149 ## read_count_as_reported_by_sra reads_downloaded ## <integer> <integer> ## SRR2443263 103977424 103977424 ## SRR2443262 125900891 125900891 ## SRR2443261 129803063 129803063 ## SRR2443260 105335395 105335395 ## SRR2443259 101692332 101692332 ## ... ... ... ## SRR2443151 87315854 87315854 ## SRR2443150 96825506 96825506 ## SRR2443148 121365435 121365435 ## SRR2443147 104038425 104038425 ## SRR2443149 113083096 113083096 ## proportion_of_reads_reported_by_sra_downloaded paired_end ## <numeric> <logical> ## SRR2443263 1 FALSE ## SRR2443262 1 FALSE ## SRR2443261 1 FALSE ## SRR2443260 1 FALSE ## SRR2443259 1 FALSE ## ... ... ... ## SRR2443151 1 FALSE ## SRR2443150 1 FALSE ## SRR2443148 1 FALSE ## SRR2443147 1 FALSE ## SRR2443149 1 FALSE ## sra_misreported_paired_end mapped_read_count auc ## <logical> <integer> <numeric> ## SRR2443263 FALSE 103499268 5149333280 ## SRR2443262 FALSE 125499809 6244059473 ## SRR2443261 FALSE 125043355 6201504759 ## SRR2443260 FALSE 104872856 5211910530 ## SRR2443259 FALSE 101258496 5033612693 ## ... ... ... ... ## SRR2443151 FALSE 86874384 4319264868 ## SRR2443150 FALSE 96316303 4787601223 ## SRR2443148 FALSE 120819733 6009515064 ## SRR2443147 FALSE 103588909 5153702232 ## SRR2443149 FALSE 112640054 5598306153 ## sharq_beta_tissue sharq_beta_cell_type ## <character> <character> ## SRR2443263 NA NA ## SRR2443262 NA NA ## SRR2443261 NA NA ## SRR2443260 NA NA ## SRR2443259 NA NA ## ... ... ... ## SRR2443151 NA NA ## SRR2443150 NA NA ## SRR2443148 NA NA ## SRR2443147 NA NA ## SRR2443149 NA NA ## biosample_submission_date biosample_publication_date ## <character> <character> ## SRR2443263 2015-08-28T16:41:29.000 2015-09-16T01:24:17.350 ## SRR2443262 2015-08-28T16:41:28.000 2015-09-16T01:24:16.410 ## SRR2443261 2015-08-28T16:41:27.000 2015-09-16T01:24:14.823 ## SRR2443260 2015-08-28T16:41:35.000 2015-09-16T01:24:13.450 ## SRR2443259 2015-08-28T16:41:33.000 2015-09-16T01:24:12.433 ## ... ... ... ## SRR2443151 2015-08-28T16:42:24.000 2015-09-16T01:19:06.787 ## SRR2443150 2015-08-28T16:42:23.000 2015-09-16T01:19:05.557 ## SRR2443148 2015-08-28T16:42:21.000 2015-09-16T01:20:16.080 ## SRR2443147 2015-08-28T16:42:19.000 2015-09-16T01:20:14.923 ## SRR2443149 2015-08-28T16:42:22.000 2015-09-16T01:19:04.583 ## biosample_update_date avg_read_length geo_accession ## <character> <integer> <character> ## SRR2443263 2015-09-16T01:28:05.297 50 GSM1863749 ## SRR2443262 2015-09-16T01:28:05.027 50 GSM1863748 ## SRR2443261 2015-09-16T01:28:04.803 50 GSM1863747 ## SRR2443260 2015-09-16T01:28:04.587 50 GSM1863746 ## SRR2443259 2015-09-16T01:28:04.347 50 GSM1863745 ## ... ... ... ... ## SRR2443151 2015-09-16T01:23:41.897 50 GSM1863637 ## SRR2443150 2015-09-16T01:23:41.453 50 GSM1863636 ## SRR2443148 2015-09-16T01:23:41.093 50 GSM1863634 ## SRR2443147 2015-09-16T01:23:40.840 50 GSM1863633 ## SRR2443149 2015-09-16T01:23:40.597 50 GSM1863635 ## bigwig_file title ## <character> <character> ## SRR2443263 SRR2443263.bw control18 ## SRR2443262 SRR2443262.bw control17 ## SRR2443261 SRR2443261.bw control16 ## SRR2443260 SRR2443260.bw control15 ## SRR2443259 SRR2443259.bw control14 ## ... ... ... ## SRR2443151 SRR2443151.bw SLE5 ## SRR2443150 SRR2443150.bw SLE4 ## SRR2443148 SRR2443148.bw SLE2 ## SRR2443147 SRR2443147.bw SLE1 ## SRR2443149 SRR2443149.bw SLE3 ## characteristics ## <CharacterList> ## SRR2443263 disease status: healthy,tissue: whole blood,anti-ro: control,... ## SRR2443262 disease status: healthy,tissue: whole blood,anti-ro: control,... ## SRR2443261 disease status: healthy,tissue: whole blood,anti-ro: control,... ## SRR2443260 disease status: healthy,tissue: whole blood,anti-ro: control,... ## SRR2443259 disease status: healthy,tissue: whole blood,anti-ro: control,... ## ... ... ## SRR2443151 disease status: systemic lupus erythematosus (SLE),tissue: whole blood,anti-ro: med,... ## SRR2443150 disease status: systemic lupus erythematosus (SLE),tissue: whole blood,anti-ro: high,... ## SRR2443148 disease status: systemic lupus erythematosus (SLE),tissue: whole blood,anti-ro: high,... ## SRR2443147 disease status: systemic lupus erythematosus (SLE),tissue: whole blood,anti-ro: high,... ## SRR2443149 disease status: systemic lupus erythematosus (SLE),tissue: whole blood,anti-ro: high,...
The most interesting part of the metadata is contained in the characteristics column, which is a CharacterList object:
colData(rse)$characteristics ## CharacterList of length 117 ## [[1]] disease status: healthy tissue: whole blood anti-ro: control ism: control ## [[2]] disease status: healthy tissue: whole blood anti-ro: control ism: control ## [[3]] disease status: healthy tissue: whole blood anti-ro: control ism: control ## [[4]] disease status: healthy tissue: whole blood anti-ro: control ism: control ## [[5]] disease status: healthy tissue: whole blood anti-ro: control ism: control ## [[6]] disease status: healthy tissue: whole blood anti-ro: control ism: control ## [[7]] disease status: healthy tissue: whole blood anti-ro: control ism: control ## [[8]] disease status: healthy tissue: whole blood anti-ro: control ism: control ## [[9]] disease status: healthy tissue: whole blood anti-ro: control ism: control ## [[10]] disease status: healthy tissue: whole blood anti-ro: control ism: control ## ... ## <107 more elements>
Let’s create some new columns with this information that can be used for the differential expression analysis. We will also make sure that they are encoded as factors and that the correct reference layer is used:
# disease status colData(rse)$disease_status <- sapply(colData(rse)$characteristics, "[", 1) colData(rse)$disease_status <- sub("disease status: ", "", colData(rse)$disease_status) colData(rse)$disease_status <- sub("systemic lupus erythematosus \\(SLE\\)", "SLE", colData(rse)$disease_status) colData(rse)$disease_status <- factor(colData(rse)$disease_status, levels = c("healthy", "SLE")) # tissue colData(rse)$tissue <- sapply(colData(rse)$characteristics, "[", 2) colData(rse)$tissue <- sub("tissue: ", "", colData(rse)$tissue) colData(rse)$tissue <- factor(colData(rse)$tissue) # anti-ro colData(rse)$anti_ro <- sapply(colData(rse)$characteristics, "[", 3) colData(rse)$anti_ro <- sub("anti-ro: ", "", colData(rse)$anti_ro) colData(rse)$anti_ro <- factor(colData(rse)$anti_ro) # ism colData(rse)$ism <- sapply(colData(rse)$characteristics, "[", 4) colData(rse)$ism <-sub("ism: ", "", colData(rse)$ism) colData(rse)$ism <- factor(colData(rse)$ism)
We can have a look at the new format:
colData(rse)[c("disease_status", "tissue", "anti_ro", "ism")] ## DataFrame with 117 rows and 4 columns ## disease_status tissue anti_ro ism ## <factor> <factor> <factor> <factor> ## SRR2443263 healthy whole blood control control ## SRR2443262 healthy whole blood control control ## SRR2443261 healthy whole blood control control ## SRR2443260 healthy whole blood control control ## SRR2443259 healthy whole blood control control ## ... ... ... ... ... ## SRR2443151 SLE whole blood med ISM_low ## SRR2443150 SLE whole blood high ISM_low ## SRR2443148 SLE whole blood high ISM_high ## SRR2443147 SLE whole blood high ISM_high ## SRR2443149 SLE whole blood high ISM_high
It looks more readable. Let’s now check how many samples we have in each group:
table(colData(rse)$disease_status) ## ## healthy SLE ## 18 99
To speed up code execution we will limit the number of SLE samples. For simplicity, we select the first 18 (healthy) and the last 18 (SLE) samples from the original RangedSummarizedExperiment object:
rse <- rse[, c(1:18, 82:99)]
Now we are ready to perform a simple differential gene expression analysis with DESeq233:
library(DESeq2) dds <- DESeqDataSet(rse, ~ disease_status) dds <- DESeq(dds) dds ## class: DESeqDataSet ## dim: 58037 36 ## metadata(1): version ## assays(5): counts mu cooks replaceCounts replaceCooks ## rownames(58037): ENSG00000000003.14 ENSG00000000005.5 ... ## ENSG00000283698.1 ENSG00000283699.1 ## rowData names(25): gene_id bp_length ... maxCooks replace ## colnames(36): SRR2443263 SRR2443262 ... SRR2443166 SRR2443165 ## colData names(27): project sample ... sizeFactor replaceable
Note that we used an extremely simple model; in the real world you will probably need to account for co-variables, potential confounders and interactions between them. edgeR34 and limma35 are good alternatives to DESEq2 for performing differential expression analyses.
We can now look at the data in more detail. We use the variance stabilising transformation (VST)36 for visualisation purposes:
vsd <- vst(dds, blind = FALSE)
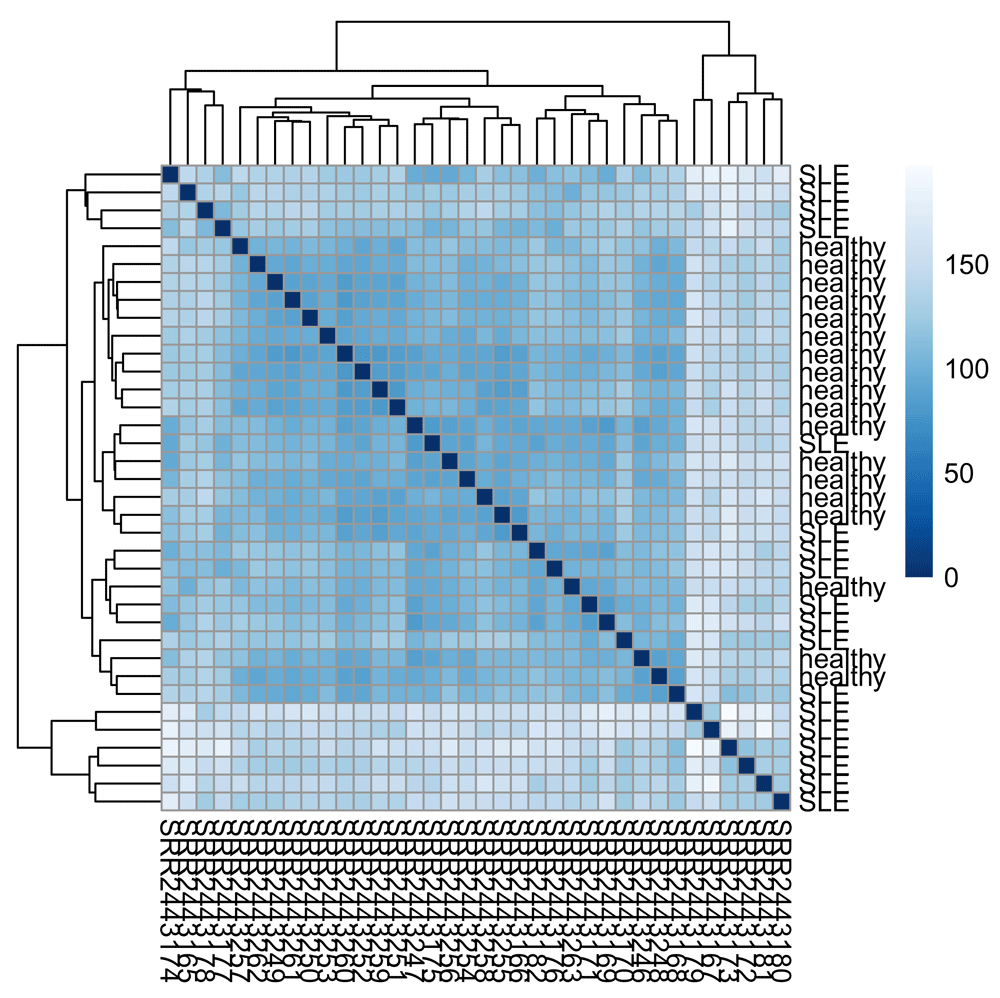
First, let’s look at distances between samples to see if we can recover a separation between SLE and healthy samples:
sampleDists <- as.matrix(dist(t(assay(vsd)))) rownames(sampleDists) <- vsd$disease_status sampleDists[c(1, 18, 19, 36), c(1, 18, 19, 36)] ## SRR2443263 SRR2443248 SRR2443182 SRR2443165 ## healthy 0.00000 106.6933 93.30292 99.84061 ## healthy 106.69330 0.0000 115.87958 127.27997 ## SLE 93.30292 115.8796 0.00000 115.06568 ## SLE 99.84061 127.2800 115.06568 0.00000
We will use the pheatmap and RColorBrewer packages for drawing the heatmap (Figure 1).
library(pheatmap) library(RColorBrewer) colors <- colorRampPalette(rev(brewer.pal(9, "Blues")))(255) pheatmap(sampleDists, col = colors)
Similarly, we can perform a principal component analysis (PCA) on the most variable 500 genes (Figure 2).
plotPCA(vsd, intgroup = "disease_status")
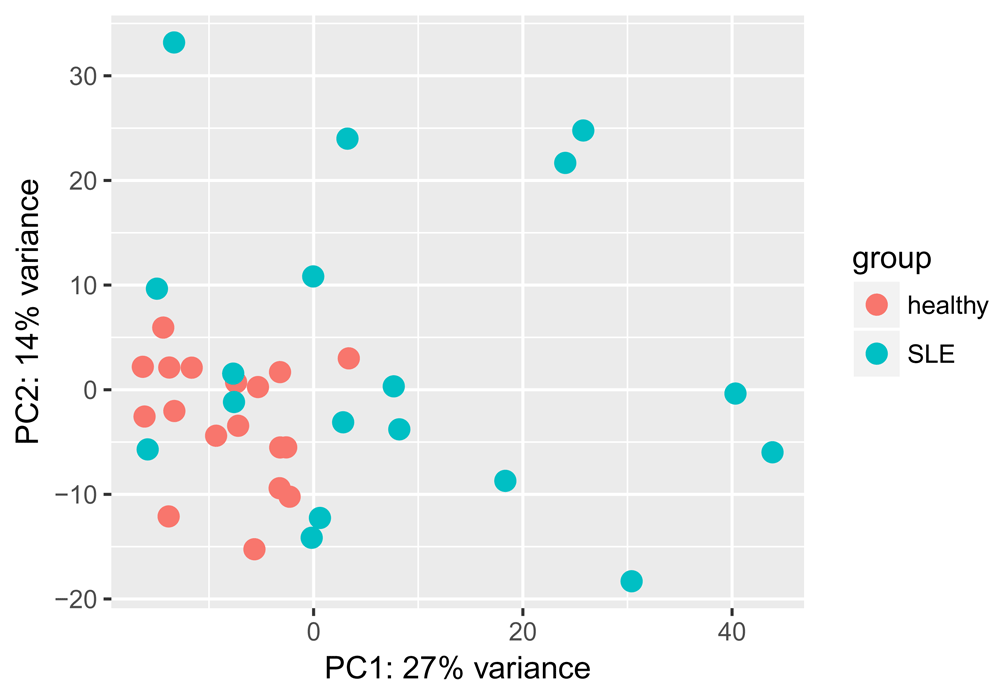
This looks better, we can see some separation of healthy and SLE samples along both PC1 and PC2, though some SLE samples appear very similar to the healthy ones. Next, we select genes that are differentially expressed below a 0.05 adjusted p-value threshold:
res <- results(dds, alpha = 0.05) res ## log2 fold change (MLE): disease status SLE vs healthy ## Wald test p-value: disease status SLE vs healthy ## DataFrame with 58037 rows and 6 columns ## baseMean log2FoldChange lfcSE stat ## <numeric> <numeric> <numeric> <numeric> ## ENSG00000000003.14 10.4189981 -0.20051804 0.24868451 -0.80631496 ## ENSG00000000005.5 0.0317823 0.03330732 2.96442394 0.01123568 ## ENSG00000000419.12 389.9025130 0.66288230 0.11427371 5.80082925 ## ENSG00000000457.13 636.6928414 0.17336365 0.08062862 2.15015047 ## ENSG00000000460.16 234.6479796 0.20589404 0.07445624 2.76530274 ## ... ... ... ... ... ## ENSG00000283695.1 0.0000000 NA NA NA ## ENSG00000283696.1 19.1311904 0.252144173 0.1545613 1.631353425 ## ENSG00000283697.1 14.9180870 0.179070242 0.1522931 1.175826692 ## ENSG00000283698.1 0.2289885 0.021962044 1.1315739 0.019408404 ## ENSG00000283699.1 0.5398951 -0.003056215 0.7578201 -0.004032903 ## pvalue padj ## <numeric> <numeric> ## ENSG00000000003.14 4.200613e-01 6.706002e-01 ## ENSG00000000005.5 9.910354e-01 NA ## ENSG00000000419.12 6.598777e-09 3.058479e-06 ## ENSG00000000457.13 3.154331e-02 1.463634e-01 ## ENSG00000000460.16 5.686999e-03 4.643041e-02 ## ... ... ... ## ENSG00000283695.1 NA NA ## ENSG00000283696.1 0.1028158 0.3075119 ## ENSG00000283697.1 0.2396641 0.4987872 ## ENSG00000283698.1 0.9845153 NA ## ENSG00000283699.1 0.9967822 NA
We can look at a summary of the results:
summary(res) ## ## out of 43005 with nonzero total read count ## adjusted p-value < 0.05 ## LFC > 0 (up) : 2526, 5.9% ## LFC < 0 (down) : 1069, 2.5% ## outliers [1] : 0, 0% ## low counts [2] : 14735, 34% ## (mean count < 1) ## [1] see 'cooksCutoff' argument of ?results ## [2] see 'independentFiltering' argument of ?results
We can also visualise the log fold changes using an MA plot (Figure 3).
plotMA(res, ylim = c(-5,5))
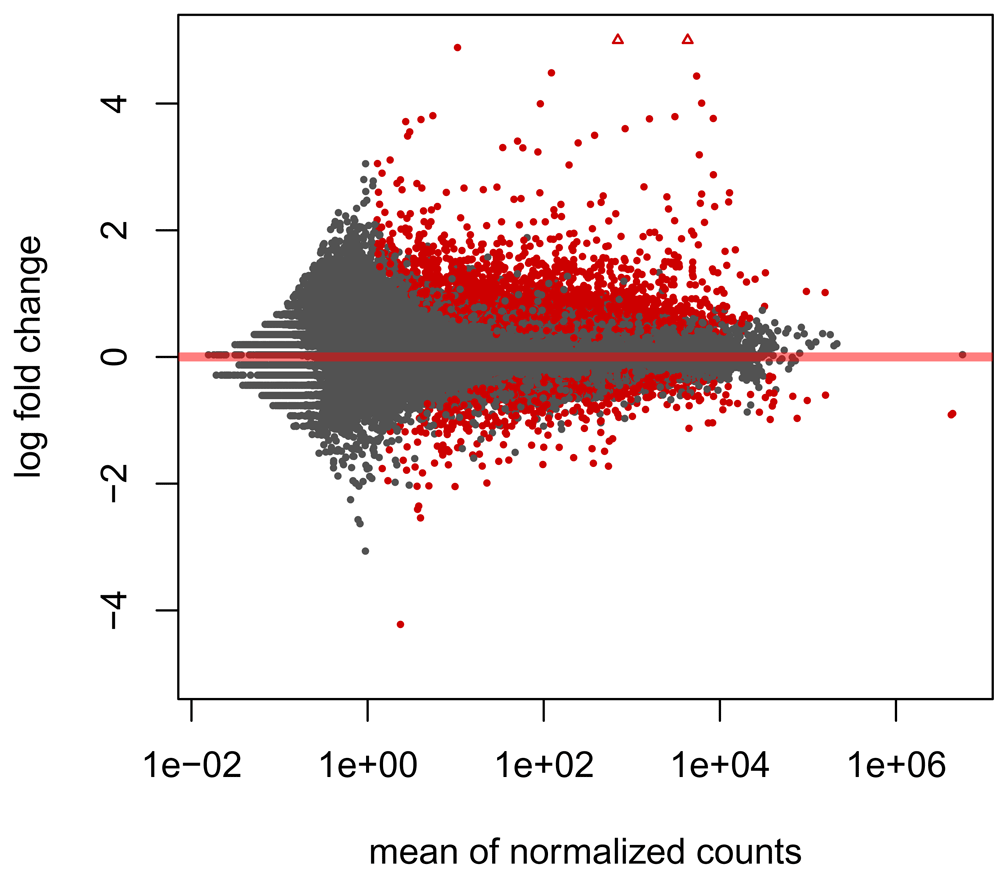
For convenience, we will save our differentially expressed genes (DEGs) in another object:
degs <- subset(res, padj < 0.05) degs <- as.data.frame(degs) head(degs) ## baseMean log2FoldChange lfcSE stat ## ENSG00000000419.12 389.90251 0.6628823 0.11427371 5.800829 ## ENSG00000000460.16 234.64798 0.2058940 0.07445624 2.765303 ## ENSG00000002549.12 1970.95648 0.8657769 0.25181202 3.438187 ## ENSG00000003096.13 11.18475 -0.7894018 0.25613621 -3.081961 ## ENSG00000003147.17 71.79432 0.6113739 0.15162606 4.032116 ## ENSG00000003249.13 119.18587 -0.8520562 0.27061961 -3.148538 ## pvalue padj ## ENSG00000000419.12 6.598777e-09 3.058479e-06 ## ENSG00000000460.16 5.686999e-03 4.643041e-02 ## ENSG00000002549.12 5.856225e-04 9.776328e-03 ## ENSG00000003096.13 2.056419e-03 2.291728e-02 ## ENSG00000003147.17 5.527679e-05 1.927054e-03 ## ENSG00000003249.13 1.640893e-03 1.955034e-02
We also map the GENCODE gene IDs to gene symbols using the annotation in the original RangedSummarizedExperiment object, which is going to be convenient later on:
rowData(rse) ## DataFrame with 58037 rows and 3 columns ## gene_id bp_length symbol ## <character> <integer> <CharacterList> ## 1 ENSG00000000003.14 4535 TSPAN6 ## 2 ENSG00000000005.5 1610 TNMD ## 3 ENSG00000000419.12 1207 DPM1 ## 4 ENSG00000000457.13 6883 SCYL3 ## 5 ENSG00000000460.16 5967 C1orf112 ## ... ... ... ... ## 58033 ENSG00000283695.1 61 NA ## 58034 ENSG00000283696.1 997 NA ## 58035 ENSG00000283697.1 1184 LOC101928917 ## 58036 ENSG00000283698.1 940 NA ## 58037 ENSG00000283699.1 60 MIR4481 degs <- merge(rowData(rse), degs, by.x = "gene_id", by.y = "row.names", all = FALSE) tail(degs) ## DataFrame with 6 rows and 9 columns ## gene_id bp_length symbol baseMean log2FoldChange ## <character> <integer> <list> <numeric> <numeric> ## [3590,] ENSG00000283444.1 831 NA 2.756993 1.3404014 ## [3591,] ENSG00000283479.1 420 NA 1.928773 1.9512651 ## [3592,] ENSG00000283485.1 2190 ASPH 277.956104 1.3415229 ## [3593,] ENSG00000283571.1 306 NA 1.791920 1.8502738 ## [3594,] ENSG00000283602.1 2089 NA 130.233552 0.5752086 ## [3595,] ENSG00000283623.1 594 ATG5 107.731105 0.4144398 ## lfcSE stat pvalue padj ## <numeric> <numeric> <numeric> <numeric> ## [3590,] 0.4729127 2.834353 0.0045918633 0.040127193 ## [3591,] 0.5681341 3.434515 0.0005936154 0.009822205 ## [3592,] 0.3694185 3.631445 0.0002818390 0.005898176 ## [3593,] 0.6557494 2.821617 0.0047782147 0.041137170 ## [3594,] 0.2047652 2.809112 0.0049678327 0.042178839 ## [3595,] 0.1066472 3.886081 0.0001018754 0.002951150
We have more than 3500 genes of interest at this stage. Since we know that therapeutic targets with genetic evidence are more likely to progress through the drug discovery pipeline6, one way to prioritise them could be to check which of these can be genetically linked to SLE. To get hold of relevant GWAS data, we will be using the gwascat Bioconductor package37, which provides an interface to the GWAS catalog38. An alternative is to use the GRASP39 database with the grasp2db40 package.
library(gwascat) # uncomment the following line to download file and build the gwasloc object all in one step #snps <- makeCurrentGwascat() # uncomment the following line to download file #download.file("http://www.ebi.ac.uk/gwas/api/search/downloads/alternative", destfile = "gwas_catalog_v1.0.1-associations_e90_r2017-12-04.tsv") snps <- read.delim("gwas_catalog_v1.0.1-associations_e90_r2017-12-04.tsv", check.names = FALSE, stringsAsFactors = FALSE) snps <- gwascat:::gwdf2GRanges(snps, extractDate = "2017-12-04") genome(snps) <- "GRCh38" snps ## gwasloc instance with 61107 records and 37 attributes per record. ## Extracted: 2017-12-04 ## Genome: GRCh38 ## Excerpt: ## GRanges object with 5 ranges and 3 metadata columns: ## seqnames ranges strand | DISEASE/TRAIT SNPS ## <Rle> <IRanges> <Rle> | <character> <character> ## [1] chr1 [203186754, 203186754] * | YKL-40 levels rs4950928 ## [2] chr13 [ 39776775, 39776775] * | Psoriasis rs7993214 ## [3] chr15 [ 78513681, 78513681] * | Lung cancer rs8034191 ## [4] chr1 [159711078, 159711078] * | Lung cancer rs2808630 ## [5] chr3 [190632672, 190632672] * | Lung cancer rs7626795 ## P-VALUE ## <numeric> ## [1] 1e-13 ## [2] 2e-06 ## [3] 3e-18 ## [4] 7e-06 ## [5] 8e-06 ## ------- ## seqinfo: 23 sequences from GRCh38 genome; no seqlengths
snps is a gwasloc object which is simply a wrapper around a GRanges object, the standard way to express genomic ranges in Bioconductor. We are interested in SNPs associated with SLE:
snps <- subsetByTraits(snps, tr = "Systemic lupus erythematosus") snps ## gwasloc instance with 402 records and 37 attributes per record. ## Extracted: 2017-12-04 ## Genome: GRCh38 ## Excerpt: ## GRanges object with 5 ranges and 3 metadata columns: ## seqnames ranges strand | ## <Rle> <IRanges> <Rle> | ## [1] chr16 [ 31301932, 31301932] * | ## [2] chr11 [ 589564, 589564] * | ## [3] chr3 [ 58384450, 58384450] * | ## [4] chr1 [173340574, 173340574] * | ## [5] chr8 [ 11491677, 11491677] * | ## DISEASE/TRAIT SNPS P-VALUE ## <character> <character> <numeric> ## [1] Systemic lupus erythematosus rs9888739 2e-23 ## [2] Systemic lupus erythematosus rs4963128 3e-10 ## [3] Systemic lupus erythematosus rs6445975 7e-09 ## [4] Systemic lupus erythematosus rs10798269 1e-07 ## [5] Systemic lupus erythematosus rs13277113 1e-10 ## ------- ## seqinfo: 23 sequences from GRCh38 genome; no seqlengths
We can visualise these as a Manhattan plot to look at the distribution of GWAS p-values over chromosomes on a negative log scale (Figure 4). Note that p-values lower than 1e-25 are truncated in the figure and that we have to load ggplot241 to modify the look of the plot:
library(ggplot2) traitsManh(gwr = snps, sel = snps, traits = "Systemic lupus erythematosus") + theme(legend.position = "none", axis.title.x = element_blank(), axis.text.x = element_blank())
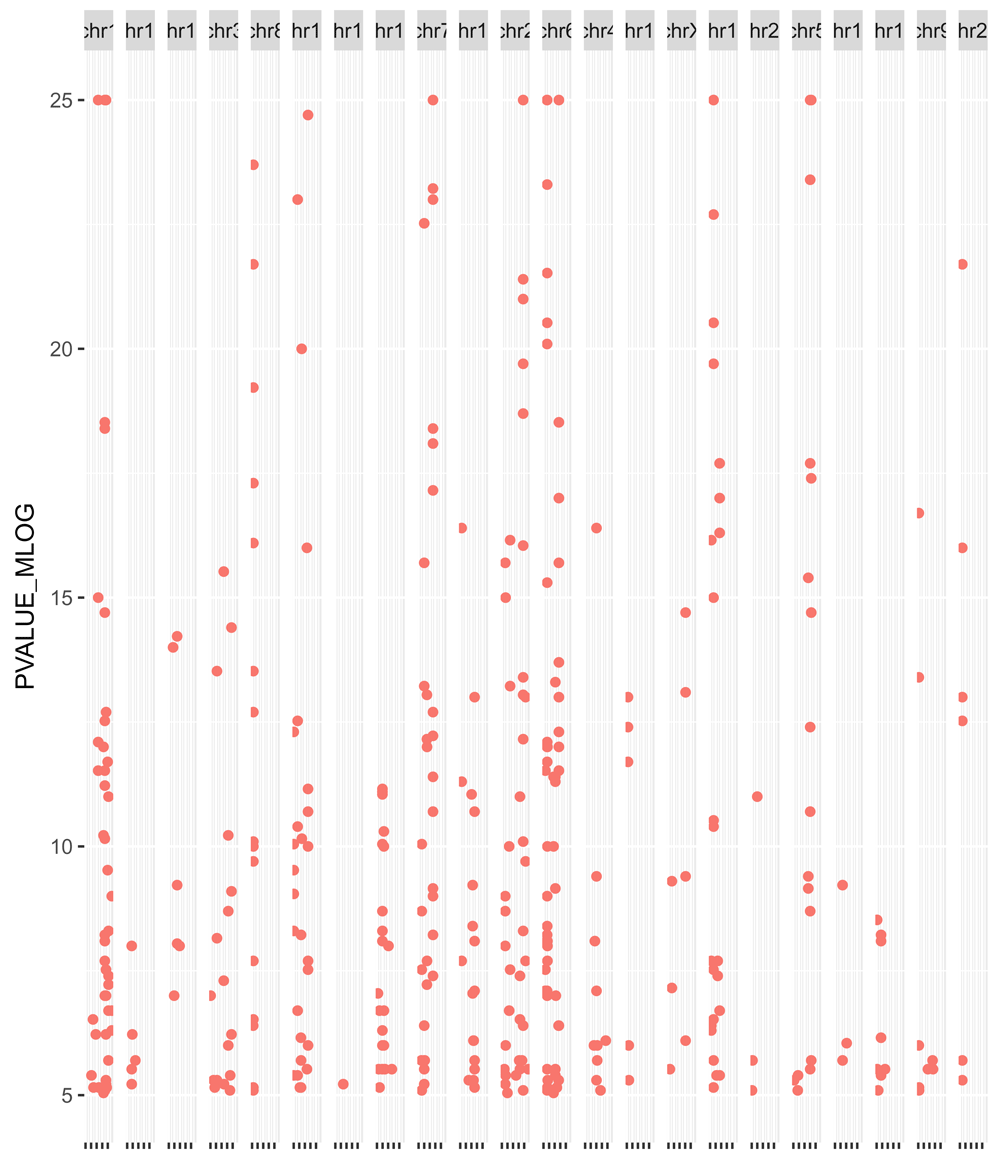
We note here that genotyping arrays typically include a very small fraction of all possible SNPs in the human genome, and there is no guarantee that the tag SNPs on the array are the true casual SNPs42. The alleles of other SNPs can be imputed from tag SNPs thanks to the structure of linkage disequilibrium (LD) blocks present in chromosomes. Thus, when linking variants to target genes in a real-world setting, it is important to take into consideration neighbouring SNPs that are in high LD and inherited with the tag SNPs. For simplicity, we will skip this LD expansion step and refer the reader to the Ensembl REST API43, the Ensembl Linkage Disequilibrium Calculator and the Bioconductor packages trio44 and ldblock45 to perform this task.
In order to annotate these variants, we need a a TxDb object, a reference of where transcripts are located on the genome. We can build this using the GenomicFeatures46 package and the GENCODE v25 gene annotation:
library(GenomicFeatures) # uncomment the following line to download file #download.file("ftp://ftp.sanger.ac.uk/pub/gencode/Gencode_human/release_25/gencode.v25.annotation.gff3.gz", destfile = "gencode.v25.annotation.gff3.gz") txdb <- makeTxDbFromGFF("gencode.v25.annotation.gff3.gz") txdb <- keepStandardChromosomes(txdb) txdb ## TxDb object: ## # Db type: TxDb ## # Supporting package: GenomicFeatures ## # Data source: gencode.v25.annotation.gff3.gz ## # Organism: NA ## # Taxonomy ID: NA ## # miRBase build ID: NA ## # Genome: NA ## # transcript_nrow: 198093 ## # exon_nrow: 1182765 ## # cds_nrow: 704859 ## # Db created by: GenomicFeatures package from Bioconductor ## # Creation time: 2018-01-18 09:02:57 +0000 (Thu, 18 Jan 2018) ## # GenomicFeatures version at creation time: 1.30.0 ## # RSQLite version at creation time: 2.0 ## # DBSCHEMAVERSION: 1.2
We also have to convert the gwasloc object into a standard GRanges object:
snps <- GRanges(snps)
Let’s check if the gwasloc and TxDb object use the same notation for chromosomes:
seqlevelsStyle(snps) ## [1] "UCSC" seqlevels(snps) ## [1] "chr1" "chr13" "chr15" "chr3" "chr8" "chr11" "chr18" "chr10" ## [9] "chr7" "chr12" "chr2" "chr6" "chr4" "chr19" "chrX" "chr16" ## [17] "chr20" "chr5" "chr14" "chr17" "chr21" "chr9" "chr22" seqlevelsStyle(txdb) ## [1] "UCSC" seqlevels(txdb) ## [1] "chr1" "chr2" "chr3" "chr4" "chr5" "chr6" "chr7" "chr8" ## [9] "chr9" "chr10" "chr11" "chr12" "chr13" "chr14" "chr15" "chr16" ## [17] "chr17" "chr18" "chr19" "chr20" "chr21" "chr22" "chrX" "chrY" ## [25] "chrM"
OK, they do. Now we can annotate our SNPs to genes using the VariantAnnotation47 package:
library(VariantAnnotation) snps_anno <- locateVariants(snps, txdb, AllVariants()) snps_anno <- unique(snps_anno) snps_anno ## GRanges object with 299 ranges and 9 metadata columns: ## seqnames ranges strand | LOCATION LOCSTART ## <Rle> <IRanges> <Rle> | <factor> <integer> ## [1] chr16 [ 31301932, 31301932] + | intron 40161 ## [2] chr11 [ 589564, 589564] + | intron 12531 ## [3] chr3 [ 58384450, 58384450] + | intron 51074 ## [4] chr1 [173340574, 173340574] * | intergenic <NA> ## [5] chr8 [ 11491677, 11491677] * | intergenic <NA> ## ... ... ... ... . ... ... ## [295] chr6 [137874014, 137874014] + | intron 6162 ## [296] chr6 [ 32619077, 32619077] * | intergenic <NA> ## [297] chr6 [137685367, 137685367] + | intron 11552 ## [298] chrX [153924366, 153924366] - | intron 1770 ## [299] chr5 [160459613, 160459613] * | intergenic <NA> ## LOCEND QUERYID TXID CDSID GENEID ## <integer> <integer> <character> <IntegerList> <character> ## [1] 40161 1 143788 ENSG00000169896.16 ## [2] 12531 2 99581 ENSG00000070047.11 ## [3] 51074 3 34101 ENSG00000168297.15 ## [4] <NA> 4 <NA> <NA> ## [5] <NA> 5 <NA> <NA> ## ... ... ... ... ... ... ## [295] 6162 393 64150 ENSG00000118503.14 ## [296] <NA> 397 <NA> <NA> ## [297] 11552 398 64145 ENSG00000230533.2 ## [298] 1770 399 196900 ENSG00000089820.15 ## [299] <NA> 400 <NA> <NA> ## PRECEDEID ## <CharacterList> ## [1] ## [2] ## [3] ## [4] ENSG00000076321.10,ENSG00000117592.8,ENSG00000117593.9,... ## [5] ENSG00000079459.12,ENSG00000136573.12,ENSG00000136574.17,... ## ... ... ## [295] ## [296] ENSG00000030110.12,ENSG00000112473.17,ENSG00000112511.17,... ## [297] ## [298] ## [299] ENSG00000118322.12,ENSG00000145864.12,ENSG00000253417.5,... ## FOLLOWID ## <CharacterList> ## [1] ## [2] ## [3] ## [4] ENSG00000094975.13,ENSG00000117560.7,ENSG00000117586.10,... ## [5] ENSG00000104643.9,ENSG00000154316.15,ENSG00000154319.14,... ## ... ... ## [295] ## [296] ENSG00000166278.14,ENSG00000168477.17,ENSG00000196126.10,... ## [297] ## [298] ## [299] ENSG00000113312.10,ENSG00000135083.14,ENSG00000145861.7,... ## ------- ## seqinfo: 23 sequences from GRCh38 genome; no seqlengths
We lost all the metadata from the original snps object, but we can recover it using the QUERYID column in snps_anno. We will only keep the SNP IDs and GWAS p-values:
snps_metadata <- snps[snps_anno$QUERYID] mcols(snps_anno) <- cbind(mcols(snps_metadata)[c("SNPS", "P-VALUE")], mcols(snps_anno)) snps_anno ## GRanges object with 299 ranges and 11 metadata columns: ## seqnames ranges strand | SNPS P.VALUE ## <Rle> <IRanges> <Rle> | <character> <numeric> ## [1] chr16 [ 31301932, 31301932] + | rs9888739 2e-23 ## [2] chr11 [ 589564, 589564] + | rs4963128 3e-10 ## [3] chr3 [ 58384450, 58384450] + | rs6445975 7e-09 ## [4] chr1 [173340574, 173340574] * | rs10798269 1e-07 ## [5] chr8 [ 11491677, 11491677] * | rs13277113 1e-10 ## ... ... ... ... . ... ... ## [295] chr6 [137874014, 137874014] + | rs5029937 5e-13 ## [296] chr6 [ 32619077, 32619077] * | rs9271366 1e-07 ## [297] chr6 [137685367, 137685367] + | rs6920220 4e-07 ## [298] chrX [153924366, 153924366] - | rs2269368 8e-07 ## [299] chr5 [160459613, 160459613] * | rs2431099 2e-06 ## LOCATION LOCSTART LOCEND QUERYID TXID CDSID ## <factor> <integer> <integer> <integer> <character> <IntegerList> ## [1] intron 40161 40161 1 143788 ## [2] intron 12531 12531 2 99581 ## [3] intron 51074 51074 3 34101 ## [4] intergenic <NA> <NA> 4 <NA> ## [5] intergenic <NA> <NA> 5 <NA> ## ... ... ... ... ... ... ... ## [295] intron 6162 6162 393 64150 ## [296] intergenic <NA> <NA> 397 <NA> ## [297] intron 11552 11552 398 64145 ## [298] intron 1770 1770 399 196900 ## [299] intergenic <NA> <NA> 400 <NA> ## GENEID ## <character> ## [1] ENSG00000169896.16 ## [2] ENSG00000070047.11 ## [3] ENSG00000168297.15 ## [4] <NA> ## [5] <NA> ## ... ... ## [295] ENSG00000118503.14 ## [296] <NA> ## [297] ENSG00000230533.2 ## [298] ENSG00000089820.15 ## [299] <NA> ## PRECEDEID ## <CharacterList> ## [1] ## [2] ## [3] ## [4] ENSG00000076321.10,ENSG00000117592.8,ENSG00000117593.9,... ## [5] ENSG00000079459.12,ENSG00000136573.12,ENSG00000136574.17,... ## ... ... ## [295] ## [296] ENSG00000030110.12,ENSG00000112473.17,ENSG00000112511.17,... ## [297] ## [298] ## [299] ENSG00000118322.12,ENSG00000145864.12,ENSG00000253417.5,... ## FOLLOWID ## <CharacterList> ## [1] ## [2] ## [3] ## [4] ENSG00000094975.13,ENSG00000117560.7,ENSG00000117586.10,... ## [5] ENSG00000104643.9,ENSG00000154316.15,ENSG00000154319.14,... ## ... ... ## [295] ## [296] ENSG00000166278.14,ENSG00000168477.17,ENSG00000196126.10,... ## [297] ## [298] ## [299] ENSG00000113312.10,ENSG00000135083.14,ENSG00000145861.7,... ## ------- ## seqinfo: 23 sequences from GRCh38 genome; no seqlengths
We can visualise where these SNPs are located with ggplot241 (Figure 5).
loc <- data.frame(table(snps_anno$LOCATION)) ggplot(data = loc, aes(x = reorder(Var1, -Freq), y = Freq)) + geom_bar(stat="identity")
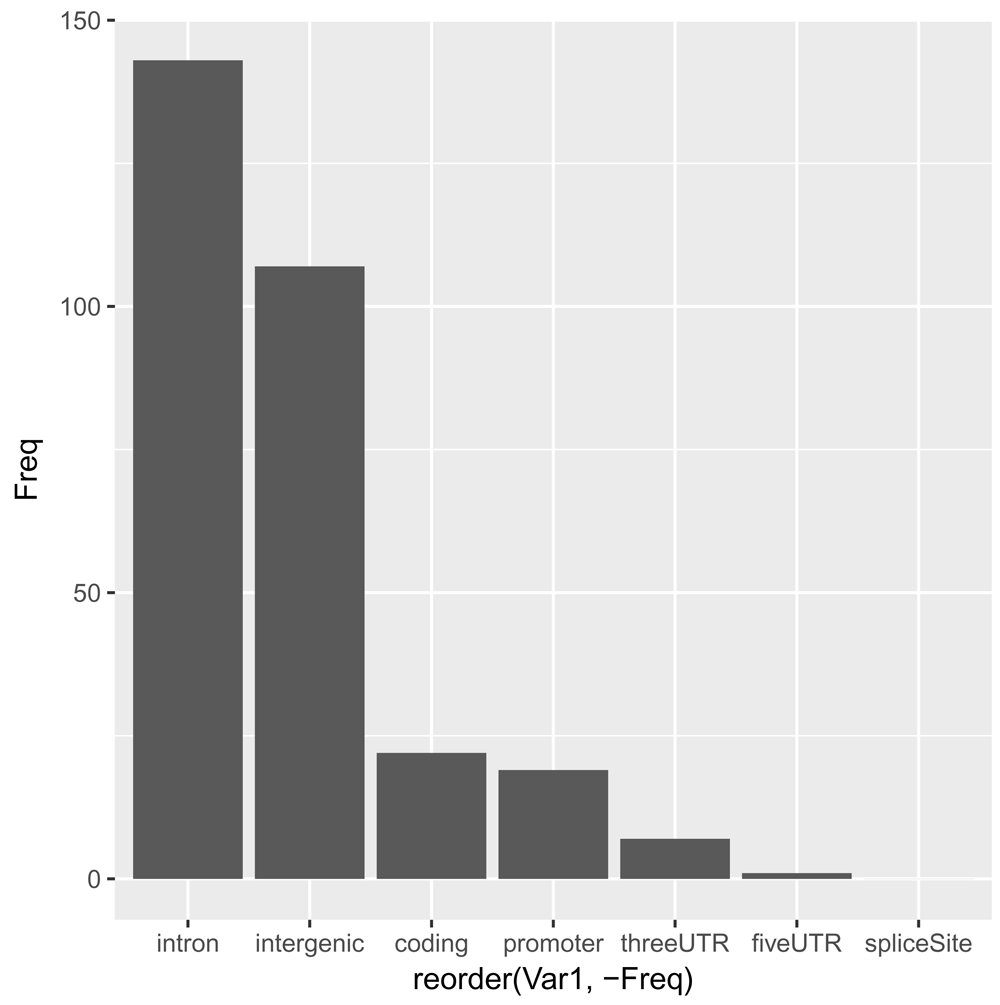
As expected11, the great majority of SNPs are located within introns and in intergenic regions. For the moment, we will focus on SNPs that are either coding or in promoter and UTR regions, as these can be assigned to target genes rather unambiguously:
snps_easy <- subset(snps_anno, LOCATION == "coding" | LOCATION == "promoter" | LOCATION == "threeUTR" | LOCATION == "fiveUTR") snps_easy <- as.data.frame(snps_easy) head(snps_easy)
## seqnames start end width strand SNPS P.VALUE LOCATION
## 1 chr4 101829919 101829919 1 + rs10516487 4e-10 coding
## 2 chr7 128954129 128954129 1 - rs10488631 2e-11 promoter
## 3 chr11 55368743 55368743 1 + rs7927370 7e-06 coding
## 4 chr6 137874929 137874929 1 + rs2230926 1e-17 coding
## 5 chr11 118702810 118702810 1 + rs4639966 1e-16 promoter
## 6 chr16 30624338 30624338 1 - rs7186852 3e-07 promoter
## LOCSTART LOCEND QUERYID TXID CDSID GENEID PRECEDEID
## 1 137 137 7 46105 170258, .... ENSG00000153064.11
## 2 NA NA 23 77786 ENSG00000275106.1
## 3 860 860 45 101610 370677 ENSG00000181958.3
## 4 380 380 57 64150 232398, .... ENSG00000118503.14
## 5 NA NA 63 104974 ENSG00000255422.1
## 6 NA NA 68 148763 ENSG00000156853.12
## FOLLOWID
## 1
## 2
## 3
## 4
## 5
## 6Now we can check if any of the genes we found to be differentially expressed in SLE is also genetically associated with the disease:
snps_easy_in_degs <- merge(degs, snps_easy, by.x = "gene_id", by.y = "GENEID", all = FALSE) snps_easy_in_degs
## DataFrame with 7 rows and 24 columns
## gene_id bp_length symbol baseMean
## <character> <integer> <list> <numeric>
## ENSG00000096968 ENSG00000096968.13 6170 JAK2 1279.47795
## ENSG00000099834 ENSG00000099834.18 3873 CDHR5 10.20177
## ENSG00000115267 ENSG00000115267.5 4528 IFIH1 1415.91330
## ENSG00000120280 ENSG00000120280.5 1855 CXorf21 637.78094
## ENSG00000185507 ENSG00000185507.19 2628 IRF7 4883.20891
## ENSG00000204366 ENSG00000204366.3 1875 ZBTB12 22.99200
## ENSG00000275106 ENSG00000275106.1 790 NA 10.32171
## log2FoldChange lfcSE stat pvalue
## <numeric> <numeric> <numeric> <numeric>
## ENSG00000096968 0.4854343 0.1553513 3.124753 1.779545e-03
## ENSG00000099834 0.8539586 0.2666557 3.202476 1.362516e-03
## ENSG00000115267 1.1494945 0.2729847 4.210838 2.544247e-05
## ENSG00000120280 0.7819504 0.1541707 5.071977 3.937038e-07
## ENSG00000185507 1.4062704 0.2992536 4.699260 2.611057e-06
## ENSG00000204366 -0.3892298 0.1348705 -2.885952 3.902318e-03
## ENSG00000275106 0.7344844 0.2305300 3.186068 1.442206e-03
## padj seqnames start end width
## <numeric> <factor> <integer> <integer> <integer>
## ENSG00000096968 2.068794e-02 chr9 4984530 4984530 1
## ENSG00000099834 1.732902e-02 chr11 625085 625085 1
## ENSG00000115267 1.120363e-03 chr2 162267541 162267541 1
## ENSG00000120280 6.047898e-05 chrX 30559729 30559729 1
## ENSG00000185507 2.298336e-04 chr11 614318 614318 1
## ENSG00000204366 3.584479e-02 chr6 31902549 31902549 1
## ENSG00000275106 1.797861e-02 chr7 128954129 128954129 1
## strand SNPS P.VALUE LOCATION LOCSTART
## <factor> <character> <numeric> <factor> <integer>
## ENSG00000096968 + rs1887428 1e-06 fiveUTR 141
## ENSG00000099834 - rs58688157 5e-13 promoter NA
## ENSG00000115267 - rs1990760 4e-08 coding 2836
## ENSG00000120280 - rs887369 5e-10 coding 627
## ENSG00000185507 - rs1061502 9e-11 coding 217
## ENSG00000204366 - rs558702 8e-21 promoter NA
## ENSG00000275106 - rs10488631 2e-11 promoter NA
## LOCEND QUERYID TXID CDSID
## <integer> <integer> <character> <list>
## ENSG00000096968 141 329 86536
## ENSG00000099834 NA 208 105793
## ENSG00000115267 2836 233 29219 106867
## ENSG00000120280 627 192 194672 692823
## ENSG00000185507 217 317 105777 385431,385427,385428,...
## ENSG00000204366 NA 116 65993
## ENSG00000275106 NA 23 77786
## PRECEDEID FOLLOWID
## <list> <list>
## ENSG00000096968
## ENSG00000099834
## ENSG00000115267
## ENSG00000120280
## ENSG00000185507
## ENSG00000204366
## ENSG00000275106So, we have 7 genes showing differential expression in SLE that are also genetically associated with the disease. While this is an interesting result, these hits are likely to be already well-known as potential SLE targets given their clear genetic association.
We will store essential information about these hits in a results data.frame:
prioritised_hits <- unique(data.frame( snp_id = snps_easy_in_degs$SNPS, snp_pvalue = snps_easy_in_degs$P.VALUE, snp_location = snps_easy_in_degs$LOCATION, gene_id = snps_easy_in_degs$gene_id, gene_symbol = snps_easy_in_degs$symbol, gene_pvalue = snps_easy_in_degs$padj, gene_log2foldchange = snps_easy_in_degs$log2FoldChange)) prioritised_hits ## snp_id snp_pvalue snp_location gene_id ## ENSG00000096968 rs1887428 1e-06 fiveUTR ENSG00000096968.13 ## ENSG00000099834 rs58688157 5e-13 promoter ENSG00000099834.18 ## ENSG00000115267 rs1990760 4e-08 coding ENSG00000115267.5 ## ENSG00000120280 rs887369 5e-10 coding ENSG00000120280.5 ## ENSG00000185507 rs1061502 9e-11 coding ENSG00000185507.19 ## ENSG00000204366 rs558702 8e-21 promoter ENSG00000204366.3 ## ENSG00000275106 rs10488631 2e-11 promoter ENSG00000275106.1 ## gene_symbol gene_pvalue gene_log2foldchange ## ENSG00000096968 JAK2 2.068794e-02 0.4854343 ## ENSG00000099834 CDHR5 1.732902e-02 0.8539586 ## ENSG00000115267 IFIH1 1.120363e-03 1.1494945 ## ENSG00000120280 CXorf21 6.047898e-05 0.7819504 ## ENSG00000185507 IRF7 2.298336e-04 1.4062704 ## ENSG00000204366 ZBTB12 3.584479e-02 -0.3892298 ## ENSG00000275106 NA 1.797861e-02 0.7344844
But what about all the SNPs in introns and intergenic regions? Some of those might be regulatory SNPs affecting the expression level of their target gene(s) through a distal enhancer. Let’s create a dataset of candidate regulatory SNPs that are either intronic or intergenic and remove the annotation obtained with VariantAnnotation:
snps_hard <- subset(snps_anno, LOCATION == "intron" | LOCATION == "intergenic", select = c("SNPS", "P.VALUE", "LOCATION")) snps_hard ## GRanges object with 250 ranges and 3 metadata columns: ## seqnames ranges strand | SNPS P.VALUE ## <Rle> <IRanges> <Rle> | <character> <numeric> ## [1] chr16 [ 31301932, 31301932] + | rs9888739 2e-23 ## [2] chr11 [ 589564, 589564] + | rs4963128 3e-10 ## [3] chr3 [ 58384450, 58384450] + | rs6445975 7e-09 ## [4] chr1 [173340574, 173340574] * | rs10798269 1e-07 ## [5] chr8 [ 11491677, 11491677] * | rs13277113 1e-10 ## ... ... ... ... . ... ... ## [246] chr6 [137874014, 137874014] + | rs5029937 5e-13 ## [247] chr6 [ 32619077, 32619077] * | rs9271366 1e-07 ## [248] chr6 [137685367, 137685367] + | rs6920220 4e-07 ## [249] chrX [153924366, 153924366] - | rs2269368 8e-07 ## [250] chr5 [160459613, 160459613] * | rs2431099 2e-06 ## LOCATION ## <factor> ## [1] intron ## [2] intron ## [3] intron ## [4] intergenic ## [5] intergenic ## ... ... ## [246] intron ## [247] intergenic ## [248] intron ## [249] intron ## [250] intergenic ## ------- ## seqinfo: 23 sequences from GRCh38 genome; no seqlengths
eQTL data. A well-established way to gain insights into target genes of regulatory SNPs is to use eQTL data, where correlations between genetic variants and expression of genes are computed across different tissues or cell types13. We will use blood eQTL data from the GTEx consortium14. To get the data, you will have to register and download the file GTEx_Analysis_v7_eQTL.tar.gz from the GTEx portal to the current working directory:
# uncomment the following line to extract the gzipped archive file #untar("GTEx_Analysis_v7_eQTL.tar.gz") gtex_blood <- read.delim(gzfile("GTEx_Analysis_v7_eQTL/Whole_Blood.v7.signif_variant_gene_pairs.txt.gz"), stringsAsFactors = FALSE) head(gtex_blood) ## variant_id gene_id tss_distance ma_samples ma_count ## 1 1_231153_CTT_C_b37 ENSG00000223972.4 219284 13 13 ## 2 1_61920_G_A_b37 ENSG00000238009.2 -67303 18 20 ## 3 1_64649_A_C_b37 ENSG00000238009.2 -64574 16 16 ## 4 1_115746_C_T_b37 ENSG00000238009.2 -13477 45 45 ## 5 1_135203_G_A_b37 ENSG00000238009.2 5980 51 51 ## 6 1_988016_T_C_b37 ENSG00000268903.1 852121 21 23 ## maf pval_nominal slope slope_se pval_nominal_threshold ## 1 0.0191740 3.69025e-08 1.319720 0.233538 1.35366e-04 ## 2 0.0281690 7.00836e-07 0.903786 0.178322 8.26088e-05 ## 3 0.0220386 5.72066e-07 1.110040 0.217225 8.26088e-05 ## 4 0.0628492 6.50297e-10 0.858203 0.134436 8.26088e-05 ## 5 0.0698630 6.67194e-10 0.811790 0.127255 8.26088e-05 ## 6 0.0318560 6.35694e-05 0.501916 0.123743 8.52870e-05 ## min_pval_nominal pval_beta ## 1 3.69025e-08 4.67848e-05 ## 2 6.50297e-10 1.11312e-06 ## 3 6.50297e-10 1.11312e-06 ## 4 6.50297e-10 1.11312e-06 ## 5 6.50297e-10 1.11312e-06 ## 6 6.35694e-05 5.44487e-02
We have to extract the genomic locations of the SNPs from the IDs used by GTEx:
locs <- strsplit(gtex_blood$variant_id, "_") gtex_blood$chr <- sapply(locs, "[", 1) gtex_blood$start <- sapply(locs, "[", 2) gtex_blood$end <- sapply(locs, "[", 2) tail(gtex_blood) ## variant_id gene_id tss_distance ma_samples ## 1052537 X_154999134_G_A_b37 ENSG00000168939.6 1660 207 ## 1052538 X_154999204_TA_T_b37 ENSG00000168939.6 1730 219 ## 1052539 X_155004280_A_G_b37 ENSG00000168939.6 6806 186 ## 1052540 X_155011926_T_C_b37 ENSG00000168939.6 14452 222 ## 1052541 X_155014420_A_G_b37 ENSG00000168939.6 16946 215 ## 1052542 X_155186978_G_C_b37 ENSG00000168939.6 189504 250 ## ma_count maf pval_nominal slope slope_se ## 1052537 259 0.351902 3.19266e-05 -0.162062 0.0383749 ## 1052538 274 0.390313 6.72752e-05 -0.157810 0.0390413 ## 1052539 224 0.303523 1.91420e-08 0.230301 0.0398809 ## 1052540 279 0.379076 3.88977e-05 0.157608 0.0377434 ## 1052541 265 0.360054 4.17781e-05 0.159699 0.0384025 ## 1052542 321 0.436141 1.24355e-04 0.145560 0.0374390 ## pval_nominal_threshold min_pval_nominal pval_beta chr start ## 1052537 0.000130368 1.9142e-08 2.75084e-05 X 154999134 ## 1052538 0.000130368 1.9142e-08 2.75084e-05 X 154999204 ## 1052539 0.000130368 1.9142e-08 2.75084e-05 X 155004280 ## 1052540 0.000130368 1.9142e-08 2.75084e-05 X 155011926 ## 1052541 0.000130368 1.9142e-08 2.75084e-05 X 155014420 ## 1052542 0.000130368 1.9142e-08 2.75084e-05 X 155186978 ## end ## 1052537 154999134 ## 1052538 154999204 ## 1052539 155004280 ## 1052540 155011926 ## 1052541 155014420 ## 1052542 155186978
We can then convert the data.frame into a GRanges object:
gtex_blood <- makeGRangesFromDataFrame(gtex_blood, keep.extra.columns = TRUE) gtex_blood ## GRanges object with 1052542 ranges and 12 metadata columns: ## seqnames ranges strand | variant_id ## <Rle> <IRanges> <Rle> | <character> ## [1] 1 [231153, 231153] * | 1_231153_CTT_C_b37 ## [2] 1 [ 61920, 61920] * | 1_61920_G_A_b37 ## [3] 1 [ 64649, 64649] * | 1_64649_A_C_b37 ## [4] 1 [115746, 115746] * | 1_115746_C_T_b37 ## [5] 1 [135203, 135203] * | 1_135203_G_A_b37 ## ... ... ... ... . ... ## [1052538] X [154999204, 154999204] * | X_154999204_TA_T_b37 ## [1052539] X [155004280, 155004280] * | X_155004280_A_G_b37 ## [1052540] X [155011926, 155011926] * | X_155011926_T_C_b37 ## [1052541] X [155014420, 155014420] * | X_155014420_A_G_b37 ## [1052542] X [155186978, 155186978] * | X_155186978_G_C_b37 ## gene_id tss_distance ma_samples ma_count maf ## <character> <integer> <integer> <integer> <numeric> ## [1] ENSG00000223972.4 219284 13 13 0.0191740 ## [2] ENSG00000238009.2 -67303 18 20 0.0281690 ## [3] ENSG00000238009.2 -64574 16 16 0.0220386 ## [4] ENSG00000238009.2 -13477 45 45 0.0628492 ## [5] ENSG00000238009.2 5980 51 51 0.0698630 ## ... ... ... ... ... ... ## [1052538] ENSG00000168939.6 1730 219 274 0.390313 ## [1052539] ENSG00000168939.6 6806 186 224 0.303523 ## [1052540] ENSG00000168939.6 14452 222 279 0.379076 ## [1052541] ENSG00000168939.6 16946 215 265 0.360054 ## [1052542] ENSG00000168939.6 189504 250 321 0.436141 ## pval_nominal slope slope_se pval_nominal_threshold ## <numeric> <numeric> <numeric> <numeric> ## [1] 3.69025e-08 1.319720 0.233538 1.35366e-04 ## [2] 7.00836e-07 0.903786 0.178322 8.26088e-05 ## [3] 5.72066e-07 1.110040 0.217225 8.26088e-05 ## [4] 6.50297e-10 0.858203 0.134436 8.26088e-05 ## [5] 6.67194e-10 0.811790 0.127255 8.26088e-05 ## ... ... ... ... ... ## [1052538] 6.72752e-05 -0.157810 0.0390413 0.000130368 ## [1052539] 1.91420e-08 0.230301 0.0398809 0.000130368 ## [1052540] 3.88977e-05 0.157608 0.0377434 0.000130368 ## [1052541] 4.17781e-05 0.159699 0.0384025 0.000130368 ## [1052542] 1.24355e-04 0.145560 0.0374390 0.000130368 ## min_pval_nominal pval_beta ## <numeric> <numeric> ## [1] 3.69025e-08 4.67848e-05 ## [2] 6.50297e-10 1.11312e-06 ## [3] 6.50297e-10 1.11312e-06 ## [4] 6.50297e-10 1.11312e-06 ## [5] 6.50297e-10 1.11312e-06 ## ... ... ... ## [1052538] 1.9142e-08 2.75084e-05 ## [1052539] 1.9142e-08 2.75084e-05 ## [1052540] 1.9142e-08 2.75084e-05 ## [1052541] 1.9142e-08 2.75084e-05 ## [1052542] 1.9142e-08 2.75084e-05 ## ------- ## seqinfo: 23 sequences from an unspecified genome; no seqlengths
We also need to ensure that the chromosome notation is consistent with the previous objects:
seqlevelsStyle(gtex_blood) ## [1] "NCBI" "Ensembl" seqlevels(gtex_blood) ## [1] "1" "2" "3" "4" "5" "6" "7" "8" "9" "10" "11" "12" "13" "14" ## [15] "15" "16" "17" "18" "19" "20" "21" "22" "X" seqlevelsStyle(gtex_blood) <- "UCSC" seqlevels(gtex_blood) ## [1] "chr1" "chr2" "chr3" "chr4" "chr5" "chr6" "chr7" "chr8" ## [9] "chr9" "chr10" "chr11" "chr12" "chr13" "chr14" "chr15" "chr16" ## [17] "chr17" "chr18" "chr19" "chr20" "chr21" "chr22" "chrX"
From the publication14, we know the genomic coordinates are mapped to genome reference GRCh37, so we will have to uplift them to GRCh38 using rtracklayer48 and a mapping (“chain”) file. The R.utils package is required to extract the gzipped file:
library(rtracklayer) library(R.utils) # uncomment the following line to download file #download.file("http://hgdownload.cse.ucsc.edu/goldenPath/hg19/liftOver/hg19T oHg38.over.chain.gz", destfile = "hg19ToHg38.over.chain.gz") # uncomment the following line to extract gzipped file #gunzip("hg19ToHg38.over.chain.gz") ch <- import.chain("hg19ToHg38.over.chain") gtex_blood <- unlist(liftOver(gtex_blood, ch))
We will use the GenomicRanges package46 to compute the overlap between GWAS SNPs and blood eQTLs:
library(GenomicRanges) hits <- findOverlaps(snps_hard, gtex_blood) snps_hard_in_gtex_blood = snps_hard[queryHits(hits)] gtex_blood_with_snps_hard = gtex_blood[subjectHits(hits)] mcols(snps_hard_in_gtex_blood) <- cbind(mcols(snps_hard_in_gtex_blood), mcols(gtex_blood_with_snps_hard)) snps_hard_in_gtex_blood <- as.data.frame(snps_hard_in_gtex_blood) head(snps_hard_in_gtex_blood) ## seqnames start end width strand SNPS P.VALUE LOCATION ## 1 chr11 589564 589564 1 + rs4963128 3e-10 intron ## 2 chr3 58384450 58384450 1 + rs6445975 7e-09 intron ## 3 chr8 11491677 11491677 1 * rs13277113 1e-10 intergenic ## 4 chr8 11491677 11491677 1 * rs13277113 1e-10 intergenic ## 5 chr8 11491677 11491677 1 * rs13277113 1e-10 intergenic ## 6 chr8 11491677 11491677 1 * rs13277113 1e-10 intergenic ## variant_id gene_id tss_distance ma_samples ma_count ## 1 11_589564_T_C_b37 ENSG00000177042.10 -105969 212 250 ## 2 3_58370177_G_T_b37 ENSG00000168291.8 -49407 205 250 ## 3 8_11349186_G_A_b37 ENSG00000154319.10 16962 157 180 ## 4 8_11349186_G_A_b37 ENSG00000136573.8 -2324 157 180 ## 5 8_11349186_G_A_b37 ENSG00000255518.1 -66284 157 180 ## 6 8_11349186_G_A_b37 ENSG00000255354.1 -68343 157 180 ## maf pval_nominal slope slope_se pval_nominal_threshold ## 1 0.339674 4.51059e-10 -0.194589 0.0301828 3.35947e-05 ## 2 0.338753 2.05231e-12 0.179408 0.0244587 6.23219e-05 ## 3 0.243902 6.46308e-27 0.778785 0.0656311 3.79430e-05 ## 4 0.243902 5.04687e-18 -0.281643 0.0305280 3.75653e-05 ## 5 0.243902 7.37464e-07 -0.262302 0.0518614 3.41126e-05 ## 6 0.243902 8.41301e-08 -0.243121 0.0442629 3.66297e-05 ## min_pval_nominal pval_beta ## 1 5.23982e-30 1.63019e-24 ## 2 3.39499e-13 3.97374e-09 ## 3 8.46904e-29 2.22416e-23 ## 4 2.97871e-19 2.22082e-14 ## 5 8.28459e-08 4.81268e-04 ## 6 2.67616e-08 1.37119e-04
So, we have 59 blood eQTL variants that are associated with SLE. We can now check whether any of the genes differentially expressed in SLE is an eGene, a gene whose expression is influenced by an eQTL. We note that gene IDs in GTEx are mapped to GENCODE v1914, while we are using the newer v25 for the DEGs. To match the gene IDs in the two objects, we will simply strip the last bit containing the GENCODE gene version, which effectively gives us Ensembl gene IDs:
snps_hard_in_gtex_blood$ensembl_id <- sub("(ENSG[0-9]+)\\.[0-9]+", "\\1", snps_hard_in_gtex_blood$gene_id) degs$ensembl_id <- sub("(ENSG[0-9]+)\\.[0-9]+", "\\1", degs$gene_id) snps_hard_in_gtex_blood_in_degs <- merge(snps_hard_in_gtex_blood, degs, by = "ensembl_id", all = FALSE) snps_hard_in_gtex_blood_in_degs ## DataFrame with 6 rows and 30 columns ## ensembl_id seqnames start end width strand ## <character> <factor> <integer> <integer> <integer> <factor> ## 1 ENSG00000130513 chr19 18370523 18370523 1 * ## 2 ENSG00000140497 chr15 75018695 75018695 1 + ## 3 ENSG00000172890 chr11 71476633 71476633 1 + ## 4 ENSG00000214894 chr6 31668965 31668965 1 + ## 5 ENSG00000214894 chr6 30973212 30973212 1 * ## 6 ENSG00000214894 chr6 31753256 31753256 1 + ## SNPS P.VALUE LOCATION variant_id gene_id.x ## <character> <numeric> <factor> <character> <character> ## 1 rs8105429 5e-06 intergenic 19_18481333_A_G_b37 ENSG00000130513.6 ## 2 rs2289583 6e-15 intron 15_75311036_C_A_b37 ENSG00000140497.12 ## 3 rs3794060 1e-20 intron 11_71187679_C_T_b37 ENSG00000172890.7 ## 4 rs9267531 8e-08 intron 6_31636742_A_G_b37 ENSG00000214894.2 ## 5 rs114090659 6e-92 intergenic 6_30940989_T_C_b37 ENSG00000214894.2 ## 6 rs3131379 2e-52 intron 6_31721033_G_A_b37 ENSG00000214894.2 ## tss_distance ma_samples ma_count maf pval_nominal slope ## <integer> <integer> <integer> <numeric> <numeric> <numeric> ## 1 -4208 166 189 0.2560980 7.87256e-11 0.350964 ## 2 145330 170 191 0.2588080 7.57250e-06 -0.107460 ## 3 23524 183 231 0.3130080 1.91380e-31 0.407266 ## 4 838306 49 54 0.0731707 3.36144e-08 0.479659 ## 5 142553 83 91 0.1233060 7.00411e-11 0.453255 ## 6 922597 50 55 0.0745257 2.69451e-08 0.479935 ## slope_se pval_nominal_threshold min_pval_nominal pval_beta ## <numeric> <numeric> <numeric> <numeric> ## 1 0.0520458 2.52102e-05 1.76820e-11 1.23175e-07 ## 2 0.0235858 6.38531e-05 2.44784e-27 1.10743e-22 ## 3 0.0310305 4.46719e-05 1.05596e-33 7.87659e-28 ## 4 0.0846154 6.02220e-05 3.17673e-13 1.77790e-08 ## 5 0.0670210 6.02220e-05 3.17673e-13 1.77790e-08 ## 6 0.0840440 6.02220e-05 3.17673e-13 1.77790e-08 ## gene_id.y bp_length symbol baseMean log2FoldChange ## <character> <integer> <list> <numeric> <numeric> ## 1 ENSG00000130513.6 2087 GDF15 6.75448 0.7883703 ## 2 ENSG00000140497.16 5000 SCAMP2 3483.03109 -0.2959934 ## 3 ENSG00000172890.11 16263 NADSYN1 4020.56224 0.2619770 ## 4 ENSG00000214894.6 2171 LINC00243 74.95034 1.2684089 ## 5 ENSG00000214894.6 2171 LINC00243 74.95034 1.2684089 ## 6 ENSG00000214894.6 2171 LINC00243 74.95034 1.2684089 ## lfcSE stat pvalue padj ## <numeric> <numeric> <numeric> <numeric> ## 1 0.28347645 2.781079 5.417861e-03 0.0448154406 ## 2 0.08814542 -3.358012 7.850510e-04 0.0119267855 ## 3 0.08976429 2.918499 3.517209e-03 0.0333810138 ## 4 0.27106143 4.679415 2.876950e-06 0.0002442643 ## 5 0.27106143 4.679415 2.876950e-06 0.0002442643 ## 6 0.27106143 4.679415 2.876950e-06 0.0002442643
We can add these 4 genes to our list:
prioritised_hits <- unique(rbind(prioritised_hits, data.frame( snp_id = snps_hard_in_gtex_blood_in_degs$SNPS, snp_pvalue = snps_hard_in_gtex_blood_in_degs$P.VALUE, snp_location = snps_hard_in_gtex_blood_in_degs$LOCATION, gene_id = snps_hard_in_gtex_blood_in_degs$gene_id.y, gene_symbol = snps_hard_in_gtex_blood_in_degs$symbol, gene_pvalue = snps_hard_in_gtex_blood_in_degs$padj, gene_log2foldchange = snps_hard_in_gtex_blood_in_degs$log2FoldChange))) prioritised_hits ## snp_id snp_pvalue snp_location gene_id ## ENSG00000096968 rs1887428 1e-06 fiveUTR ENSG00000096968.13 ## ENSG00000099834 rs58688157 5e-13 promoter ENSG00000099834.18 ## ENSG00000115267 rs1990760 4e-08 coding ENSG00000115267.5 ## ENSG00000120280 rs887369 5e-10 coding ENSG00000120280.5 ## ENSG00000185507 rs1061502 9e-11 coding ENSG00000185507.19 ## ENSG00000204366 rs558702 8e-21 promoter ENSG00000204366.3 ## ENSG00000275106 rs10488631 2e-11 promoter ENSG00000275106.1 ## 1 rs8105429 5e-06 intergenic ENSG00000130513.6 ## 2 rs2289583 6e-15 intron ENSG00000140497.16 ## 3 rs3794060 1e-20 intron ENSG00000172890.11 ## 4 rs9267531 8e-08 intron ENSG00000214894.6 ## 5 rs114090659 6e-92 intergenic ENSG00000214894.6 ## 6 rs3131379 2e-52 intron ENSG00000214894.6 ## gene_symbol gene_pvalue gene_log2foldchange ## ENSG00000096968 JAK2 2.068794e-02 0.4854343 ## ENSG00000099834 CDHR5 1.732902e-02 0.8539586 ## ENSG00000115267 IFIH1 1.120363e-03 1.1494945 ## ENSG00000120280 CXorf21 6.047898e-05 0.7819504 ## ENSG00000185507 IRF7 2.298336e-04 1.4062704 ## ENSG00000204366 ZBTB12 3.584479e-02 -0.3892298 ## ENSG00000275106 NA 1.797861e-02 0.7344844 ## 1 GDF15 4.481544e-02 0.7883703 ## 2 SCAMP2 1.192679e-02 -0.2959934 ## 3 NADSYN1 3.338101e-02 0.2619770 ## 4 LINC00243 2.442643e-04 1.2684089 ## 5 LINC00243 2.442643e-04 1.2684089 ## 6 LINC00243 2.442643e-04 1.2684089
FANTOM5 data. The FANTOM consortium profiled gene expression across a large panel of tissues and cell types using CAGE19,21. This technology allows mapping of transcription start sites (TSSs) and enhancer RNAs (eRNAs) genome-wide. Correlations between these promoter and enhancer elements across a large panel of tissues and cell types can then be calculated to identify significant promoter - enhancer pairs. In turn, we will use these correlations to map distal regulatory SNPs to target genes.
We can read in and have a look at the enhancer - promoter correlation data in this way:
# uncomment the following line to download the file #download.file("http://enhancer.binf.ku.dk/presets/enhancer_tss_associations.bed", destfile = "enhancer_tss_associations.bed") fantom <- read.delim("enhancer_tss_associations.bed", skip = 1, stringsAsFactors = FALSE) head(fantom) ## X.chrom chromStart chromEnd ## 1 chr1 858252 861621 ## 2 chr1 894178 956888 ## 3 chr1 901376 956888 ## 4 chr1 901376 1173762 ## 5 chr1 935051 942164 ## 6 chr1 935051 1005621 ## name ## 1 chr1:858256-858648;NM_152486;SAMD11;R:0.404;FDR:0 ## 2 chr1:956563-956812;NM_015658;NOC2L;R:0.202;FDR:8.01154668254404e-08 ## 3 chr1:956563-956812;NM_001160184,NM_032129;PLEKHN1;R:0.422;FDR:0 ## 4 chr1:1173386-1173736;NM_001160184,NM_032129;PLEKHN1;R:0.311;FDR:0 ## 5 chr1:941791-942135;NM_001142467,NM_021170;HES4;R:0.187;FDR:6.32949888009368e-07 ## 6 chr1:1005293-1005547;NM_001142467,NM_021170;HES4;R:0.236;FDR:6.28221217150423e-11 ## score strand thickStart thickEnd itemRgb blockCount blockSizes ## 1 404 . 858452 858453 0,0,0 2 401,1001 ## 2 202 . 956687 956688 0,0,0 2 1001,401 ## 3 422 . 956687 956688 0,0,0 2 1001,401 ## 4 311 . 1173561 1173562 0,0,0 2 1001,401 ## 5 187 . 941963 941964 0,0,0 2 1001,401 ## 6 236 . 1005420 1005421 0,0,0 2 1001,401 ## chromStarts ## 1 0,2368 ## 2 0,62309 ## 3 0,55111 ## 4 0,271985 ## 5 0,6712 ## 6 0,70169
Everything we need is in the fourth column, name: genomic location of the enhancer, gene identifiers, Pearson correlation coefficient and significance. We will use the splitstackshape package to parse it:
library(splitstackshape) fantom <- as.data.frame(cSplit(fantom, splitCols = "name", sep = ";", direction = "wide")) head(fantom) ## X.chrom chromStart chromEnd score strand thickStart thickEnd itemRgb ## 1 chr1 858252 861621 404 . 858452 858453 0,0,0 ## 2 chr1 894178 956888 202 . 956687 956688 0,0,0 ## 3 chr1 901376 956888 422 . 956687 956688 0,0,0 ## 4 chr1 901376 1173762 311 . 1173561 1173562 0,0,0 ## 5 chr1 935051 942164 187 . 941963 941964 0,0,0 ## 6 chr1 935051 1005621 236 . 1005420 1005421 0,0,0 ## blockCount blockSizes chromStarts name_1 ## 1 2 401,1001 0,2368 chr1:858256-858648 ## 2 2 1001,401 0,62309 chr1:956563-956812 ## 3 2 1001,401 0,55111 chr1:956563-956812 ## 4 2 1001,401 0,271985 chr1:1173386-1173736 ## 5 2 1001,401 0,6712 chr1:941791-942135 ## 6 2 1001,401 0,70169 chr1:1005293-1005547 ## name_2 name_3 name_4 name_5 ## 1 NM_152486 SAMD11 R:0.404 FDR:0 ## 2 NM_015658 NOC2L R:0.202 FDR:8.01154668254404e-08 ## 3 NM_001160184,NM_032129 PLEKHN1 R:0.422 FDR:0 ## 4 NM_001160184,NM_032129 PLEKHN1 R:0.311 FDR:0 ## 5 NM_001142467,NM_021170 HES4 R:0.187 FDR:6.32949888009368e-07 ## 6 NM_001142467,NM_021170 HES4 R:0.236 FDR:6.28221217150423e-11
Now we can extract the genomic locations of the enhancers and the correlation values:
locs <- strsplit(as.character(fantom$name_1), "[:-]") fantom$chr <- sapply(locs, "[", 1) fantom$start <- as.numeric(sapply(locs, "[", 2)) fantom$end <- as.numeric(sapply(locs, "[", 3)) fantom$symbol <- fantom$name_3 fantom$corr <- sub("R:", "", fantom$name_4) fantom$fdr <- sub("FDR:", "", fantom$name_5) head(fantom) ## X.chrom chromStart chromEnd score strand thickStart thickEnd itemRgb ## 1 chr1 858252 861621 404 . 858452 858453 0,0,0 ## 2 chr1 894178 956888 202 . 956687 956688 0,0,0 ## 3 chr1 901376 956888 422 . 956687 956688 0,0,0 ## 4 chr1 901376 1173762 311 . 1173561 1173562 0,0,0 ## 5 chr1 935051 942164 187 . 941963 941964 0,0,0 ## 6 chr1 935051 1005621 236 . 1005420 1005421 0,0,0 ## blockCount blockSizes chromStarts name_1 ## 1 2 401,1001 0,2368 chr1:858256-858648 ## 2 2 1001,401 0,62309 chr1:956563-956812 ## 3 2 1001,401 0,55111 chr1:956563-956812 ## 4 2 1001,401 0,271985 chr1:1173386-1173736 ## 5 2 1001,401 0,6712 chr1:941791-942135 ## 6 2 1001,401 0,70169 chr1:1005293-1005547 ## name_2 name_3 name_4 name_5 chr ## 1 NM_152486 SAMD11 R:0.404 FDR:0 chr1 ## 2 NM_015658 NOC2L R:0.202 FDR:8.01154668254404e-08 chr1 ## 3 NM_001160184,NM_032129 PLEKHN1 R:0.422 FDR:0 chr1 ## 4 NM_001160184,NM_032129 PLEKHN1 R:0.311 FDR:0 chr1 ## 5 NM_001142467,NM_021170 HES4 R:0.187 FDR:6.32949888009368e-07 chr1 ## 6 NM_001142467,NM_021170 HES4 R:0.236 FDR:6.28221217150423e-11 chr1 ## start end symbol corr fdr ## 1 858256 858648 SAMD11 0.404 0 ## 2 956563 956812 NOC2L 0.202 8.01154668254404e-08 ## 3 956563 956812 PLEKHN1 0.422 0 ## 4 1173386 1173736 PLEKHN1 0.311 0 ## 5 941791 942135 HES4 0.187 6.32949888009368e-07 ## 6 1005293 1005547 HES4 0.236 6.28221217150423e-11
We can select only the enhancer - promoter pairs with a decent level of correlation and significance and tidy the data at the same time:
fantom <- unique(subset(fantom, subset = corr >= 0.25 & fdr < 1e-5, select = c("chr", "start", "end", "symbol"))) head(fantom) ## chr start end symbol ## 1 chr1 858256 858648 SAMD11 ## 3 chr1 956563 956812 PLEKHN1 ## 4 chr1 1173386 1173736 PLEKHN1 ## 13 chr1 1136075 1136463 ISG15 ## 14 chr1 956563 956812 AGRN ## 27 chr1 1060905 1061095 RNF223
Now we would like to check whether any of our candidate regulatory SNPs are falling in any of these enhancers. To do this, we have to convert the data.frame into a GRanges object:
fantom <- makeGRangesFromDataFrame(fantom, keep.extra.columns = TRUE) fantom ## GRanges object with 33957 ranges and 1 metadata column: ## seqnames ranges strand | symbol ## <Rle> <IRanges> <Rle> | <factor> ## 1 chr1 [ 858256, 858648] * | SAMD11 ## 3 chr1 [ 956563, 956812] * | PLEKHN1 ## 4 chr1 [1173386, 1173736] * | PLEKHN1 ## 13 chr1 [1136075, 1136463] * | ISG15 ## 14 chr1 [ 956563, 956812] * | AGRN ## ... ... ... ... . ... ## 66929 chrX [154256125, 154256514] * | F8A2 ## 66932 chrY [ 2871660, 2871926] * | ZFY ## 66933 chrY [ 2872046, 2872325] * | ZFY ## 66940 chrY [ 21664138, 21664302] * | KDM5D ## 66941 chrY [ 22735456, 22735677] * | EIF1AY ## ------- ## seqinfo: 24 sequences from an unspecified genome; no seqlengths
Similar to the GTEx data, the FANTOM5 data is also mapped to GRCh3719, so we will have to uplift the GRCh37 coordinates to GRCh38:
fantom <- unlist(liftOver(fantom, ch)) fantom ## GRanges object with 34160 ranges and 1 metadata column: ## seqnames ranges strand | symbol ## <Rle> <IRanges> <Rle> | <factor> ## 1 chr1 [ 922876, 923268] * | SAMD11 ## 3 chr1 [1021183, 1021432] * | PLEKHN1 ## 4 chr1 [1238006, 1238356] * | PLEKHN1 ## 13 chr1 [1200695, 1201083] * | ISG15 ## 14 chr1 [1021183, 1021432] * | AGRN ## ... ... ... ... . ... ## 66929 chrX [155027850, 155028239] * | F8A2 ## 66932 chrY [ 3003619, 3003885] * | ZFY ## 66933 chrY [ 3004005, 3004284] * | ZFY ## 66940 chrY [ 19502252, 19502416] * | KDM5D ## 66941 chrY [ 20573570, 20573791] * | EIF1AY ## ------- ## seqinfo: 24 sequences from an unspecified genome; no seqlengths
We can now compute the overlap between SNPs and enhancers:
hits <- findOverlaps(snps_hard, fantom) snps_hard_in_fantom = snps_hard[queryHits(hits)] fantom_with_snps_hard = fantom[subjectHits(hits)] mcols(snps_hard_in_fantom) <- cbind(mcols(snps_hard_in_fantom), mcols(fantom_with_snps_hard)) snps_hard_in_fantom <- as.data.frame(snps_hard_in_fantom) snps_hard_in_fantom ## seqnames start end width strand SNPS P.VALUE LOCATION ## 1 chr2 191099907 191099907 1 - rs7574865 9e-14 intron ## 2 chr2 191099907 191099907 1 - rs7574865 9e-14 intron ## 3 chr6 32082981 32082981 1 - rs1150754 6e-29 intron ## 4 chr6 32082981 32082981 1 - rs1150754 6e-29 intron ## 5 chr6 32082981 32082981 1 - rs1150754 6e-29 intron ## 6 chr6 32689659 32689659 1 * rs3129716 4e-09 intergenic ## 7 chr6 32689659 32689659 1 * rs3129716 4e-09 intergenic ## 8 chr6 32689659 32689659 1 * rs3129716 4e-09 intergenic ## 9 chr6 32689659 32689659 1 * rs3129716 4e-09 intergenic ## 10 chr6 32689659 32689659 1 * rs3129716 4e-09 intergenic ## 11 chr6 32689659 32689659 1 * rs3129716 4e-09 intergenic ## 12 chr1 235876577 235876577 1 - rs9782955 1e-09 intron ## 13 chr7 50267214 50267214 1 * rs11185603 4e-07 intergenic ## 14 chr11 73152652 73152652 1 * rs11235667 7e-11 intergenic ## symbol ## 1 NAB1 ## 2 STAT4 ## 3 LY6G6C ## 4 TNXB ## 5 PPT2 ## 6 HLA-DQB1 ## 7 HLA-DOB ## 8 HLA-DMA ## 9 HLA-DOA ## 10 HLA-DPA1 ## 11 HLA-DPB1 ## 12 LYST ## 13 IKZF1 ## 14 FCHSD2
We note that some of the SNPs are assigned to more than one gene. This is because enhancers are promiscuous and can regulate multiple genes.
We can now check if any of these genes is differentially expressed in our RNA-seq data:
snps_hard_in_fantom_in_degs <- merge(snps_hard_in_fantom, degs, by = "symbol", all = FALSE) snps_hard_in_fantom_in_degs ## DataFrame with 2 rows and 18 columns ## symbol seqnames start end width strand SNPS ## <factor> <factor> <integer> <integer> <integer> <factor> <character> ## 1 HLA-DOA chr6 32689659 32689659 1 * rs3129716 ## 2 IKZF1 chr7 50267214 50267214 1 * rs11185603 ## P.VALUE LOCATION gene_id bp_length baseMean ## <numeric> <factor> <character> <integer> <numeric> ## 1 4e-09 intergenic ENSG00000204252.13 4012 962.7578 ## 2 4e-07 intergenic ENSG00000185811.16 9784 7183.7639 ## log2FoldChange lfcSE stat pvalue padj ## <numeric> <numeric> <numeric> <numeric> <numeric> ## 1 -0.4424595 0.15882236 -2.785877 0.0053383163 0.04431304 ## 2 -0.2575717 0.07647486 -3.368057 0.0007569983 0.01162554 ## ensembl_id ## <character> ## 1 ENSG00000204252 ## 2 ENSG00000185811
We have identified 2 genes whose putative enhancers contain SLE GWAS SNPs. Let’s add these to our list:
prioritised_hits <- unique(rbind(prioritised_hits, data.frame( snp_id = snps_hard_in_fantom_in_degs$SNPS, snp_pvalue = snps_hard_in_fantom_in_degs$P.VALUE, snp_location = snps_hard_in_fantom_in_degs$LOCATION, gene_id = snps_hard_in_fantom_in_degs$gene_id, gene_symbol = snps_hard_in_fantom_in_degs$symbol, gene_pvalue = snps_hard_in_fantom_in_degs$padj, gene_log2foldchange = snps_hard_in_fantom_in_degs$log2FoldChange))) prioritised_hits ## snp_id snp_pvalue snp_location gene_id ## ENSG00000096968 rs1887428 1e-06 fiveUTR ENSG00000096968.13 ## ENSG00000099834 rs58688157 5e-13 promoter ENSG00000099834.18 ## ENSG00000115267 rs1990760 4e-08 coding ENSG00000115267.5 ## ENSG00000120280 rs887369 5e-10 coding ENSG00000120280.5 ## ENSG00000185507 rs1061502 9e-11 coding ENSG00000185507.19 ## ENSG00000204366 rs558702 8e-21 promoter ENSG00000204366.3 ## ENSG00000275106 rs10488631 2e-11 promoter ENSG00000275106.1 ## 1 rs8105429 5e-06 intergenic ENSG00000130513.6 ## 2 rs2289583 6e-15 intron ENSG00000140497.16 ## 3 rs3794060 1e-20 intron ENSG00000172890.11 ## 4 rs9267531 8e-08 intron ENSG00000214894.6 ## 5 rs114090659 6e-92 intergenic ENSG00000214894.6 ## 6 rs3131379 2e-52 intron ENSG00000214894.6 ## 11 rs3129716 4e-09 intergenic ENSG00000204252.13 ## 21 rs11185603 4e-07 intergenic ENSG00000185811.16 ## gene_symbol gene_pvalue gene_log2foldchange ## ENSG00000096968 JAK2 2.068794e-02 0.4854343 ## ENSG00000099834 CDHR5 1.732902e-02 0.8539586 ## ENSG00000115267 IFIH1 1.120363e-03 1.1494945 ## ENSG00000120280 CXorf21 6.047898e-05 0.7819504 ## ENSG00000185507 IRF7 2.298336e-04 1.4062704 ## ENSG00000204366 ZBTB12 3.584479e-02 -0.3892298 ## ENSG00000275106 NA 1.797861e-02 0.7344844 ## 1 GDF15 4.481544e-02 0.7883703 ## 2 SCAMP2 1.192679e-02 -0.2959934 ## 3 NADSYN1 3.338101e-02 0.2619770 ## 4 LINC00243 2.442643e-04 1.2684089 ## 5 LINC00243 2.442643e-04 1.2684089 ## 6 LINC00243 2.442643e-04 1.2684089 ## 11 HLA-DOA 4.431304e-02 -0.4424595 ## 21 IKZF1 1.162554e-02 -0.2575717
Promoter Capture Hi-C data. More recently, chromatin interaction data was generated across 17 human primary blood cell types25. More than 30,000 promoter baits were used to capture promoter-interacting regions genome-wide. These regions were then mapped to enhancers based on the Ensembl Regulatory Build49 and can be accessed in the supplementary data of the paper:
# uncomment the following line to download file #download.file("http://www.cell.com/cms/attachment/2086554122/2074217047/mmc4 .zip", destfile = "mmc4.zip") # uncomment the following lines to extract zipped files #unzip("mmc4.zip") #unzip("DATA_S1.zip") pchic <- read.delim("ActivePromoterEnhancerLinks.tsv", stringsAsFactors = FALSE) head(pchic) ## baitChr baitSt baitEnd baitID oeChr oeSt oeEnd oeID ## 1 chr1 1206873 1212438 254 chr1 943676 957199 228 ## 2 chr1 1206873 1212438 254 chr1 1034268 1040208 235 ## 3 chr1 1206873 1212438 254 chr1 1040208 1043143 236 ## 4 chr1 1206873 1212438 254 chr1 1069045 1083958 242 ## 5 chr1 1206873 1212438 254 chr1 1083958 1091234 243 ## 6 chr1 1206873 1212438 254 chr1 1585571 1619752 304 ## cellType.s. ## 1 nCD8 ## 2 nCD4,nCD8,Mac0,Mac1,Mac2,MK,Mon ## 3 nCD4,nCD8,Mac0,Mac1,Mac2,MK ## 4 nCD8 ## 5 nCD8 ## 6 Neu ## sample.s. ## 1 C0066PH1 ## 2 S007DDH2,S007G7H4,C0066PH1,S00C2FH1,S00390H1,S001MJH1,S001S7H2,S0022IH2,S0062 2H1,S00BS4H1,S004BTH2,C000S5H2 ## 3 S007DDH2,S007G7H4,C0066PH1,S00C2FH1,S00390H1,S001MJH1,S001S7H2,S0022IH2,S0062 2H1,S00BS4H1,S004BTH2 ## 4 C0066PH1,S00C2FH1 ## 5 C0066PH1,S00C2FH1 ## 6 C000S5H1
In this case, we will have to map the promoter baits to genes first. We can do this by converting the baits to a GRanges object and then using the TxDb object we previously built to extract positions of transcription start sites (TSSs):
baits <- GRanges(seqnames = pchic$baitChr, ranges = IRanges(start = pchic$baitSt, end = pchic$baitEnd)) tsss <- promoters(txdb, upstream = 0, downstream = 1, columns = "gene_id") hits <- nearest(baits, tsss) baits$gene_id <- unlist(tsss[hits]$gene_id) baits ## GRanges object with 51142 ranges and 1 metadata column: ## seqnames ranges strand | gene_id ## <Rle> <IRanges> <Rle> | <character> ## [1] chr1 [1206873, 1212438] * | ENSG00000186827.10 ## [2] chr1 [1206873, 1212438] * | ENSG00000186827.10 ## [3] chr1 [1206873, 1212438] * | ENSG00000186827.10 ## [4] chr1 [1206873, 1212438] * | ENSG00000186827.10 ## [5] chr1 [1206873, 1212438] * | ENSG00000186827.10 ## ... ... ... ... . ... ## [51138] chrY [22732049, 22743996] * | ENSG00000230727.1 ## [51139] chrY [22732049, 22743996] * | ENSG00000230727.1 ## [51140] chrY [22732049, 22743996] * | ENSG00000230727.1 ## [51141] chrY [22732049, 22743996] * | ENSG00000230727.1 ## [51142] chrY [22732049, 22743996] * | ENSG00000230727.1 ## ------- ## seqinfo: 24 sequences from an unspecified genome; no seqlengths
Now we can create a GRanges object of the enhancers in the promoter capture Hi-C data with the bait annotation attached:
pchic <- GRanges(seqnames = pchic$oeChr, ranges = IRanges(start = pchic$oeSt, end = pchic$oeEnd), gene_id = baits$gene_id) pchic <- unique(pchic) pchic ## GRanges object with 25232 ranges and 1 metadata column: ## seqnames ranges strand | gene_id ## <Rle> <IRanges> <Rle> | <character> ## [1] chr1 [ 943676, 957199] * | ENSG00000186827.10 ## [2] chr1 [1034268, 1040208] * | ENSG00000186827.10 ## [3] chr1 [1040208, 1043143] * | ENSG00000186827.10 ## [4] chr1 [1069045, 1083958] * | ENSG00000186827.10 ## [5] chr1 [1083958, 1091234] * | ENSG00000186827.10 ## ... ... ... ... . ... ## [25228] chrY [23401616, 23404873] * | ENSG00000230727.1 ## [25229] chrY [23404938, 23407193] * | ENSG00000230727.1 ## [25230] chrY [23409014, 23410287] * | ENSG00000230727.1 ## [25231] chrY [23410287, 23411837] * | ENSG00000230727.1 ## [25232] chrY [23411837, 23412539] * | ENSG00000230727.1 ## ------- ## seqinfo: 24 sequences from an unspecified genome; no seqlengths
Next, we basically repeat the steps we have taken when working with the FANTOM5 data to find SLE GWAS SNPs overlapping with these enhancers:
hits <- findOverlaps(snps_hard, pchic) snps_hard_in_pchic = snps_hard[queryHits(hits)] pchic_with_snps_hard = pchic[subjectHits(hits)] mcols(snps_hard_in_pchic) <- cbind(mcols(snps_hard_in_pchic), mcols(pchic_with_snps_hard)) snps_hard_in_pchic <- as.data.frame(snps_hard_in_pchic) snps_hard_in_pchic ## seqnames start end width strand SNPS P.VALUE ## 1 chr6 31753256 31753256 1 + rs3131379 2e-52 ## 2 chr6 32696681 32696681 1 * rs2647012 8e-06 ## 3 chr16 30631546 30631546 1 * rs7197475 3e-08 ## 4 chr20 4762059 4762059 1 * rs6084875 2e-06 ## 5 chr6 32689659 32689659 1 * rs3129716 4e-09 ## 6 chr6 31668965 31668965 1 + rs9267531 8e-08 ## 7 chr6 31951083 31951083 1 + rs1270942 2e-165 ## 8 chr6 106140931 106140931 1 - rs6568431 5e-14 ## 9 chr7 28146272 28146272 1 - rs849142 9e-11 ## 10 chr2 65381229 65381229 1 - rs268134 1e-10 ## 11 chr17 39850937 39850937 1 - rs143123127 6e-09 ## 12 chr9 86916761 86916761 1 * rs190029011 3e-06 ## 13 chr11 65637829 65637829 1 * rs931127 7e-06 ## 14 chr19 18370523 18370523 1 * rs8105429 5e-06 ## 15 chr16 85977731 85977731 1 * rs10521318 4e-06 ## 16 chr5 39406395 39406395 1 - rs3914167 8e-06 ## 17 chr16 31315385 31315385 1 + rs11860650 2e-20 ## LOCATION gene_id ## 1 intron ENSG00000219797.2 ## 2 intergenic ENSG00000204290.10 ## 3 intergenic ENSG00000180096.11 ## 4 intergenic ENSG00000212536.1 ## 5 intergenic ENSG00000204290.10 ## 6 intron ENSG00000219797.2 ## 7 intron ENSG00000225851.1 ## 8 intron ENSG00000112297.14 ## 9 intron ENSG00000106052.13 ## 10 intron ENSG00000198369.9 ## 11 intron ENSG00000277363.4 ## 12 intergenic ENSG00000223012.1 ## 13 intergenic ENSG00000245532.6 ## 14 intergenic ENSG00000099308.10 ## 15 intergenic ENSG00000279907.1 ## 16 intron ENSG00000212296.1 ## 17 intron ENSG00000260060.1
We check if any of these enhancers containing SLE variants are known to putatively regulate genes differentially expressed in SLE:
snps_hard_in_pchic_in_degs <- merge(snps_hard_in_pchic, degs, by = "gene_id", all = FALSE) snps_hard_in_pchic_in_degs ## DataFrame with 4 rows and 18 columns ## gene_id seqnames start end width strand ## <character> <factor> <integer> <integer> <integer> <factor> ## 1 ENSG00000106052.13 chr7 28146272 28146272 1 - ## 2 ENSG00000219797.2 chr6 31753256 31753256 1 + ## 3 ENSG00000219797.2 chr6 31668965 31668965 1 + ## 4 ENSG00000245532.6 chr11 65637829 65637829 1 * ## SNPS P.VALUE LOCATION bp_length symbol baseMean ## <character> <numeric> <factor> <integer> <list> <numeric> ## 1 rs849142 9e-11 intron 9165 TAX1BP1 2406.26093 ## 2 rs3131379 2e-52 intron 498 NA 74.58175 ## 3 rs9267531 8e-08 intron 498 NA 74.58175 ## 4 rs931127 7e-06 intergenic 22767 NEAT1,MIR612 17580.27601 ## log2FoldChange lfcSE stat pvalue padj ## <numeric> <numeric> <numeric> <numeric> <numeric> ## 1 0.3438396 0.1205716 2.851746 4.347982e-03 0.0386695506 ## 2 0.5586633 0.1116884 5.001982 5.674388e-07 0.0000798169 ## 3 0.5586633 0.1116884 5.001982 5.674388e-07 0.0000798169 ## 4 0.5259525 0.1366133 3.849935 1.181492e-04 0.0032213554 ## ensembl_id ## <character> ## 1 ENSG00000106052 ## 2 ENSG00000219797 ## 3 ENSG00000219797 ## 4 ENSG00000245532
And finally we add these 3 genes to our list. These are our final results:
prioritised_hits <- unique(rbind(prioritised_hits, data.frame( snp_id = snps_hard_in_pchic_in_degs$SNPS, snp_pvalue = snps_hard_in_pchic_in_degs$P.VALUE, snp_location = snps_hard_in_pchic_in_degs$LOCATION, gene_id = snps_hard_in_pchic_in_degs$gene_id, gene_symbol = snps_hard_in_pchic_in_degs$symbol, gene_pvalue = snps_hard_in_pchic_in_degs$padj, gene_log2foldchange = snps_hard_in_pchic_in_degs$log2FoldChange))) prioritised_hits ## snp_id snp_pvalue snp_location gene_id ## ENSG00000096968 rs1887428 1e-06 fiveUTR ENSG00000096968.13 ## ENSG00000099834 rs58688157 5e-13 promoter ENSG00000099834.18 ## ENSG00000115267 rs1990760 4e-08 coding ENSG00000115267.5 ## ENSG00000120280 rs887369 5e-10 coding ENSG00000120280.5 ## ENSG00000185507 rs1061502 9e-11 coding ENSG00000185507.19 ## ENSG00000204366 rs558702 8e-21 promoter ENSG00000204366.3 ## ENSG00000275106 rs10488631 2e-11 promoter ENSG00000275106.1 ## 1 rs8105429 5e-06 intergenic ENSG00000130513.6 ## 2 rs2289583 6e-15 intron ENSG00000140497.16 ## 3 rs3794060 1e-20 intron ENSG00000172890.11 ## 4 rs9267531 8e-08 intron ENSG00000214894.6 ## 5 rs114090659 6e-92 intergenic ENSG00000214894.6 ## 6 rs3131379 2e-52 intron ENSG00000214894.6 ## 11 rs3129716 4e-09 intergenic ENSG00000204252.13 ## 21 rs11185603 4e-07 intergenic ENSG00000185811.16 ## 12 rs849142 9e-11 intron ENSG00000106052.13 ## 22 rs3131379 2e-52 intron ENSG00000219797.2 ## 31 rs9267531 8e-08 intron ENSG00000219797.2 ## 41 rs931127 7e-06 intergenic ENSG00000245532.6 ## gene_symbol gene_pvalue gene_log2foldchange ## ENSG00000096968 JAK2 2.068794e-02 0.4854343 ## ENSG00000099834 CDHR5 1.732902e-02 0.8539586 ## ENSG00000115267 IFIH1 1.120363e-03 1.1494945 ## ENSG00000120280 CXorf21 6.047898e-05 0.7819504 ## ENSG00000185507 IRF7 2.298336e-04 1.4062704 ## ENSG00000204366 ZBTB12 3.584479e-02 -0.3892298 ## ENSG00000275106 NA 1.797861e-02 0.7344844 ## 1 GDF15 4.481544e-02 0.7883703 ## 2 SCAMP2 1.192679e-02 -0.2959934 ## 3 NADSYN1 3.338101e-02 0.2619770 ## 4 LINC00243 2.442643e-04 1.2684089 ## 5 LINC00243 2.442643e-04 1.2684089 ## 6 LINC00243 2.442643e-04 1.2684089 ## 11 HLA-DOA 4.431304e-02 -0.4424595 ## 21 IKZF1 1.162554e-02 -0.2575717 ## 12 TAX1BP1 3.866955e-02 0.3438396 ## 22 NA 7.981690e-05 0.5586633 ## 31 NA 7.981690e-05 0.5586633 ## 41 NEAT1, M.... 3.221355e-03 0.5259525
In this Bioconductor workflow we have used several packages and datasets to demonstrate how regulatory genomic data can be used to annotate significant hits from GWASs and provide an intermediate layer connecting genetics and transcriptomics. Overall, we identified 17 SLE-associated SNPs that we mapped to 16 genes differentially expressed in SLE, using eQTL data14 and enhancer - promoter relationships from CAGE19 and promoter capture Hi-C experiments25.
While simplified, the workflow also demonstrates some real-world challenges encountered when working with genomic data from different sources, such as the use of different genome references and gene annotation conventions, the parsing of files with custom formats into Bioconductor-compatible objects and the mapping of genomic locations to genes.
As the sample size and power of GWASs and gene expression studies continue to increase, it will become more and more challenging to identify truly significant hits and interpret them. The use of regulatory genomics data as presented here can be an important skill and tool to gain insights into large biomedical datasets and help in the identification of biomarkers and therapeutic targets.
Download links for all datasets are part of the workflow. Software packages required to reproduce the analysis can be installed as part of the workflow. Code is available at https://github.com/enricoferrero/bioconductor-regulatory-genomics-workflow.
Archived code as at time of publication: http://doi.org/10.5281/zenodo.115423550
License: CC-BY 4.0
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)