Keywords
Dystrophin, DAPC, Duchenne muscular dystrophy, Smooth muscle, Blood vessels, Neuregulin, ERBB2/4, HER2/4
Dystrophin, DAPC, Duchenne muscular dystrophy, Smooth muscle, Blood vessels, Neuregulin, ERBB2/4, HER2/4
Signaling from neuregulin 1 (NRG), through its epidermal growth factor (EGF) family of receptors ERBB1-4, has major functions in several organs such as heart, breast, and nervous system including central and peripheral synapses. The role of NRG signaling in skeletal muscle has been controversial. To investigate signaling events in muscle fibers, myotubes formed by fusion of myoblasts, are routinely used as the in vitro equivalent of muscle fibers. We already reported that in myotubes, formed from C2C12 myoblasts, NRG signaling through ERBB2/4 heterodimeric receptors phosphorylated α-dystrobrevin 1 (α-DB1)1, one of the components of the dystrophin associated protein complex (DAPC). DAPC links the intracellular actin cytoskeleton to the extracellular matrix and is thereby thought to provide structural stability during muscular activity. DAPC, apart from containing dystrophin which is a 427 kDa protein, consists of several other proteins such as α- and β-dystrobrevins, dystroglycans, sarcoglycans, sarcospan, syntrophins, and laminins. At the neuromuscular synapse, the DAPC is also formed with utrophin, also a 427 kDa protein, instead of dystrophin. The phosphorylation of α-DB1 through NRG/ERBB signaling stabilized acetylcholine receptors (AChRs) at the neuromuscular synapse2. Duchenne and Becker muscular dystrophy (D/BMD) patients have mutation(s) in the dystrophin gene, resulting in the expression of a truncated dystrophin protein3–6. Taken together, the main body of research on DMD argues for the lack of dystrophin in skeletal muscle as the cause for DMD.
In mice, apart from muscular dystrophy, absence of dystrophin causes neuromuscular junction (NMJ) fragmentation7 similar to the NMJ fragmentation associated with a loss of NRG/ERBB signaling1. Lack of dystrophin, besides causing muscular dystrophies, results in cardiomyopathy8 and is also responsible for several disease states in the brain6. The importance of NRG signaling for normal cardiac development in mice was firmly established by the fact that ablation of NRG, ERBB4, or ERBB2 resulted in premature death during midgestation9–11. In cardiac muscle NRG/ERBB4 signaling is sufficient for cardiomyocyte proliferation and repair of heart injury12, but knowledge of the detailed signaling mechanisms and the target proteins through which this was achieved are lacking. The aim of this investigation was to identify the function of NRG/ERBB signaling in muscle and, as it phosphorylated α-DB1 in the DAPC complex, determine if it had a functional role in muscular dystrophy by identifying downstream signaling targets.
erbb2/4 dKO and loxP flanked erbb2/4 myoblasts (a kind gift from M. Courtet, and previously described1) were cultured on laminin-coated dishes (Roche) and upon reaching 70–80% confluency, were allowed to form myotubes by changing to differentiation media (2% horse serum, 1% penicillin/streptomycin (Sigma-Aldrich), DMEM (Sigma-Aldrich)).
Myotubes from 10-cm culture dishes were harvested in 600 μl lysis buffer and protein complexes were immunoprecipitated as described previously1,13 with modifications. In brief, myotubes harvested in ice-cold lysis buffer (10 mM Na3PO4, pH 7.8, 150 mM NaCl, 5 mM EDTA, 1 mM EGTA, 1% Triton X-100, protease inhibitor mixture (Roche), and phosphatase inhibitors Pic1 and Pic2 (Sigma-Aldrich)) were homogenized in a Dounce homogenizer and incubated for 3 h at 4°C with protein G-coupled mouse monoclonal syntrophin antibody 1351 (4 μl/80 μl protein G beads, Abcam, catalog number ab11425). Beads were then washed in lysis buffer containing protease inhibitors, but without Triton X-100, resuspended in 3 x SDS loading buffer (150 mM Tris-HCl [pH 6.8], 300 mM dithiothreitol [added just before use], 6% SDS, 0.3% bromophenol blue, and 30% glycerol), and denatured (94°C, 3 min) before loading on an 6–8% gradient/0.8% bis-acrylamide SDS-PAGE gels buffered with Tris-glycine.
Gels were transferred onto PVDF membranes (Millipore) and subject to ECL (Thermo Fisher Scientific) development after incubation with primary and secondary antibodies. BSA (3%) was used as a blocking reagent. The following primary antibodies were used: rabbit anti-dystrophin (H300) polyclonal (diluted 1:400, catalog number sc-15376) and mouse anti-utrophin (55) monoclonal (diluted 1:400, catalog number sc-136116) were from Santa Cruz Biotechnology, Inc., mouse monoclonal anti-syntrophin 1351 (4 μl antibody/80 μl protein G beads for lysate from a 10 cm culture dish of myotubes, catalog number ab11425) from abcam and rabbit anti-α-syntrophin 259 (5 μg/ml for Westerns; a kind gift from Stanly C. Froehner and Marvin Adams, University of Washington, Seattle, WA). Goat anti-mouse IgG-HRP (catalog number sc-2005) and goat anti-rabbit IgG-HRP (catalog number sc-2004) secondary antibodies (Santa Cruz Biotechnology, Inc.) were used at a 1:5,000 dilution.
RNA isolation and qPCR were performed as previously described14 and the 2−ΔΔCt method was used to analyze relative changes in gene expression. RNA from myotube cultures was isolated with TRIzol (Invitrogen) according to their protocol. DNase I (Promega) treatment and reverse transcription was performed on 1 µg total RNA with random primers and superscript reverse transcriptase from Invitrogen according to their protocol. cDNA was diluted 1:5 before use in qPCR, which was performed with SyBR Green mix (Applied Biosystems) using the Applied Biosystems StepOne machine with two-step PCR (60°C, 1 min and 95°C 15 s) for 40 cycles using the standard program. The quantitative PCR mix was prepared as follows: 12.5 μl SyBR Green mix, 2.5 μl of a 3 µM solution each of forward and reverse primer, 1 μl of diluted cDNA and made up to 25 µl total volume with sterile water. Each sample for real time PCR was done in triplicate and the mean of the resulting three values were taken. The following primers, designed to recognize exons with at least one intron in between for each primer pair, were used for dystrophin, utrophin, and rL8 amplifications: dystrophin forward, 5′-GATGATGAACATTTGTTAATCCAGC-3′ and reverse, 5′-CATATTCTGCTTGCAGATTCCTG-3′; utrophin forward, 5′-CTAAACTCCTGCGGCAGCAC-3′ and reverse, '-GTGTCAAGTGAGTAGCTCAATGC-3′ and rL8 (normalization gene) forward, 5′-ACTGGACAGTTCGTGTACTG-3′ and reverse, 5′-GCTTCACTCGAGTCTTCTTG-3′.
We reported previously that on western blots following immunoprecipitation, there were two isoforms of α-dystrobrevin1 associated with DAPC, a 75 kDa and 89 kDa protein1. We also demonstrated that ablation of ERBB2/4 receptors resulted in a lack of phosphorylation of the 75 kDa protein1. In addition, the amount of the 75 kDa protein detected on western blots, compared to the 89 kDa protein, was reduced in Erbb2/4 dKO myotubes. However in myotubes with intact ERBB2/4 receptors, such as in C2C12 myotubes, it was the other way around1 i.e. less 89 kDa protein. As absence of dystrophin in the DMD mouse model mdx, also resulted in a reduction in the 75 kDa protein compared to wild-type mice15, it was possible that the reduction in the 75 kDa protein in the Erbb2/4 dKO myotubes was due to a reduction in dystrophin. Furthermore the AChR fragmentation observed in mdx mice7, paralleled those seen in Erbb2/4 dKO mice1 raising the possibility that another reason for the observed destabilization of the AChR cluster in Erbb2/4 dKO myotubes could be due to the reduced levels of dystrophin in these myotubes. These observations taken together suggest that one of the targets of NRG/ERBB signaling is dystrophin. To investigate this, Erbb2/4 dKO myotubes derived from immortalized Erbb2/4 dKO myoblasts1 and myotubes formed from myoblasts, before transfection of myoblasts with Cre recombinase, containing loxP flanked Erbb2/4 genes were used. Both, ERBB2 and ERBB4 receptors were ablated to eliminate NRG signaling1 through these receptors in muscle, because ERBB4 receptors, apart from forming heterodimers, can also form homodimers and ERBB2 can heterodimerize with ERBB316. Furthermore, as cultured myotubes secrete NRG17, the external addition of NRG was not necessary, as demonstrated for phosphorylation of α-dystrobrevin 1 by NRG/ERBB1.
Three independent experiments (Figure 1A) each using myotubes formed from a different myoblast clone containing loxP-flanked Erbb2/4 genes, clearly detected dystrophin (lanes 1–3). However dystrophin was not detected in Erbb2/4 dKO myotubes (lanes 4–6) where ERBB2/4 receptors were ablated after CRE recombination of loxP flanked Erbb2/4 genes. Utrophin on the other hand was detected in myotubes with and without ERBB2/4 receptors (Figure 1B) demonstrating that NRG/ERBB signaling selectively regulates dystrophin expression (Figure 2).
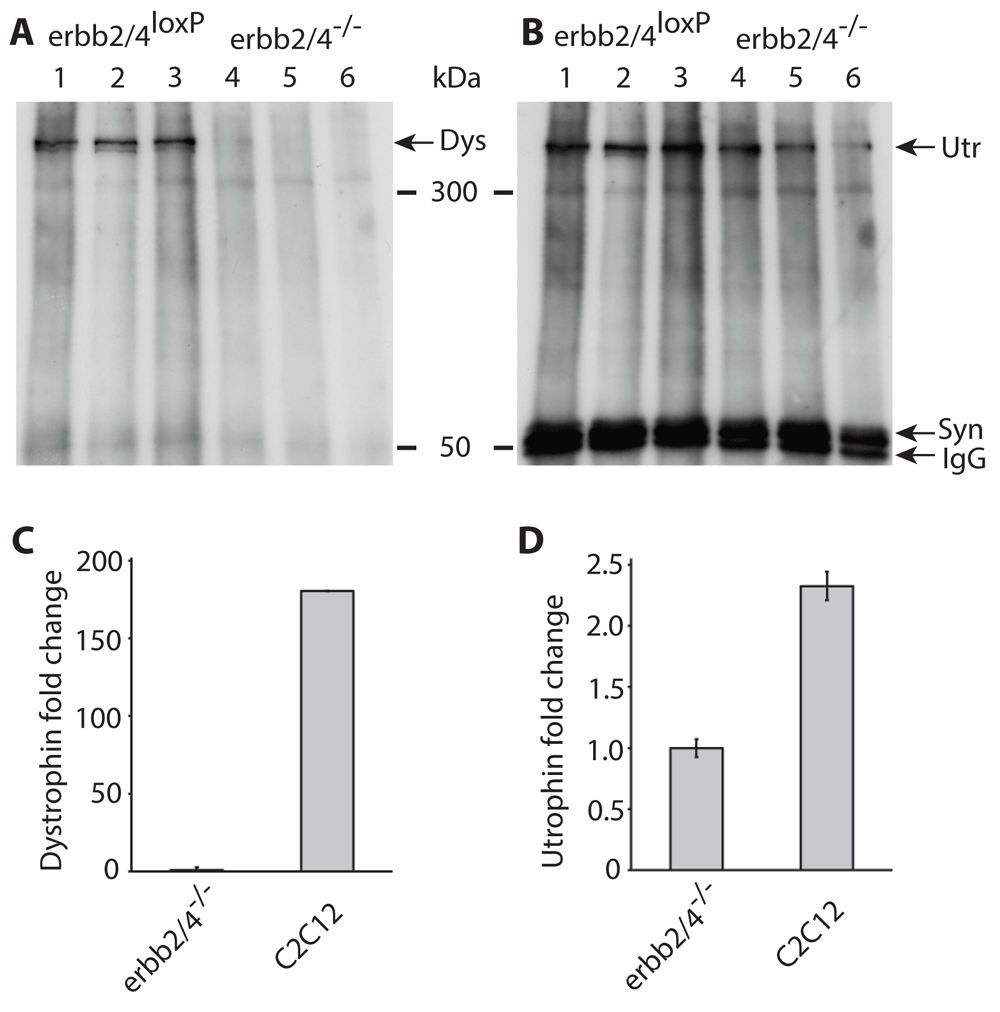
(A and B), Western blots of immunoprecipitated proteins from myotubes with loxP flanked exons of Erbb2 and Erbb4 gene (lanes 1 to 3) and cre mediated knock-out of Erbb2 and Erbb4 genes (lanes 4 to 6) detected with dystrophin (A) and utrophin (B) antibodies. Lanes 1 to 3 and 4 to 6 each represent the same experiment performed independently and loaded on the same gel. The lower panel shows detection of syntrophin that served as a loading control. Immunoglobulin G (IgG) detected is the syntrophin antibody used for the immunoprecipitation. As the western blot in (A) was stripped of antibodies and used in (B), the loading control in (B) applies to both A and B. (C and D) qPCR data of dystrophin and utrophin levels in C2C12 myotubes, relative to Erbb2/4 dKO (Erbb2/4-/-) myotubes. Expression levels were normalized to ribosomal protein L8 (rL8) expression. This experiment was performed at least twice with similar results. This figure was previously published in a patent (Patent Link: WO 2017/036852 A1), but the copyright is the author’s own.
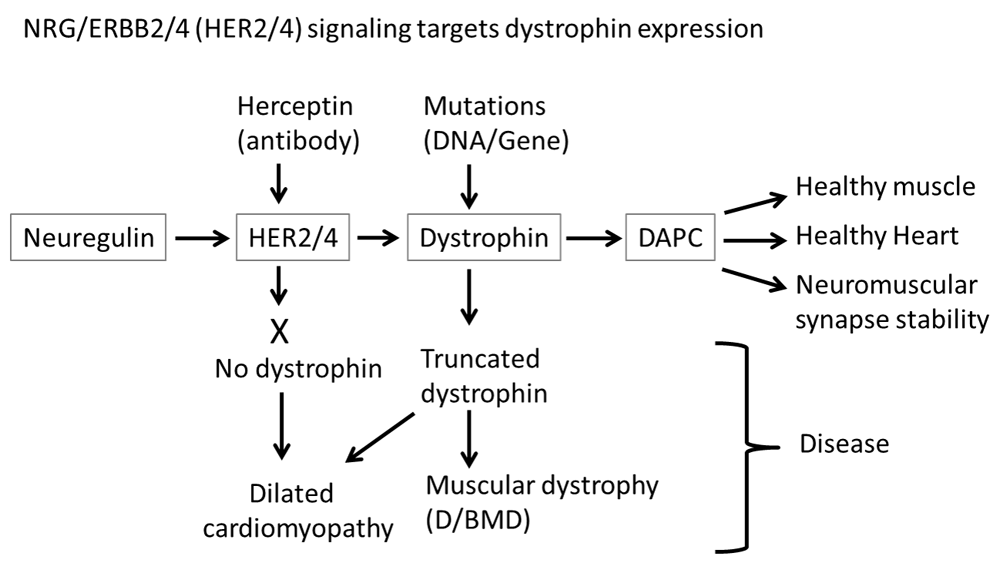
Under normal circumstances, NRG signaling through ERBB2/4 (HER2/4) receptors stimulates dystrophin expression, allowing the formation of a normal dystrophin-associated protein complex (DAPC). If either the dystrophin gene contains mutations or NRG signaling is blocked, then in the absence of functional dystrophin, a normal DAPC is not formed, resulting in various disease states such as dilated cardiomyopathy, Duchenne/Becker muscular dystrophy (D/BMD), and neuromuscular synapse instability.
The qPCR estimation of dystrophin and utrophin levels (Figure 1C, D) confirmed that dystrophin expression is absent in Erbb2/4 dKO (shown in Figure 1 as erbb2/4-/-) myotubes, as there was no detectable mRNA and signals in qPCR were only observed above threshold at about 34 cycles which is essentially detection of non-specific amplification or background signal, whereas detection of ribosomal protein L8, used to normalize expression of dystrophin and utrophin, was above threshold at about 22 cycles in Erbb2/4 dKO and C2C12 samples. Detection of dystrophin mRNA in C2C12 cells by qPCR confirmed that the primers used to amplify dystrophin functioned. It is only possible to conclude that dystrophin is present in C2C12 and the level cannot be estimated since the level of dystrophin in C2C12 was relative to that in Erbb2/4 dKO myotubes, for which essentially background non-specific values were obtained in qPCR due to the absence of dystrophin mRNA. Utrophin detection (Figure 1D), using the same RNA/cDNA preparation used for dystrophin detection, confirmed that the cDNA preparation from Erbb2/4 dKO myotubes used for dystrophin detection was intact. Utrophin expression was reduced to only less than half the amount (Figure 1D) in Erbb2/4 dKO myotubes compared to C2C12 myotubes which may be due to the lack of NRG signaling since NRG stimulates utrophin expression to some extent18. Myotubes formed from C2C12 myoblasts were used as a control for qPCR instead of myotubes containing loxP flanked Erbb2/4 genes (used for the western blots in Figure 1A, B) as the loxP-flanked Erbb4 gene19 is a hypomorph due to the insertion of the neo selection cassette. Hence C2C12 myotubes were used instead of Erbb2/4 dKO myotubes to exclude the possibility that levels of dystrophin mRNA may have been affected (NRG signals through ERBB2/4 to stimulate dystrophin expression and reduced Erbb4 expression may have affected this). This is not a problem for dystrophin protein detection (not estimation) in immunoprecipitated samples from myotubes containing loxP flanked Erbb2/4 genes. qPCR on Erbb2/4 dKO confirmed the absence of dystrophin expression (Figure 1C) as observed on the western blot (Figure 1A). Hence NRG/ERBB signaling is necessary for dystrophin expression.
Even though dystrophin is lacking in skeletal muscles of Erbb2/4 dKO mice, they do not show dystrophic symptoms20. The promoter used for CRE expression in generating Erbb2/4 dKO mice, the human skeletal actin promoter (HSA), is expressed in the striated muscles, skeletal and heart muscle15,21,22. Therefore ERBB2/4 and dystrophin levels in smooth muscle of blood vessels would not be affected, as CRE is not expressed in smooth muscle of Erbb2/4 dKO mice, allowing the formation of a normal functional DAPC. Hence smooth muscle of blood vessels in these mice allows for increased blood flow to skeletal and cardiac muscle during exercise. This is because neuronal nitric oxide synthase (nNOS) associating with dystrophin and generating nitric oxide (NO) that signals to soluble guanylate cyclase, generating cyclic guanosine 3’,5’-monophosphate (cGMP) in smooth muscle of blood vessels, causes vasodilation enabling exercise-induced increase of blood flow and thereby prevents muscle ischemia23. Thus the absence of obvious dystrophic symptoms in Erbb2/4 dKO skeletal muscle where there is a lack of dystrophin strongly suggests that the main cause of muscular dystrophy is not the lack of dystrophin in skeletal muscle per se but systemic lack of functional dystrophin, especially in smooth muscle of blood vessels, resulting in impaired sympatholysis and muscle ischemia during exercise23. This hypothesis is consistent with published data using phosphodiesterase type 5 (PDE5) inhibitors, which interfere with breakdown of NO by PDE5 and thereby prolong the half-life of cGMP, the target of NO24. This was demonstrated in mdx mice where PDE5 inhibition alleviates the dystrophic phenotype23 and also in DMD patients where PDE5 inhibition with either tadalafil or sildenafil treatment in Duchenne muscular dystrophy boys restored normal blood vessel function and blood flow during exercise24. Thus the muscle has an extensive vasculature to provide more oxygenated blood through vasodilation the absence of which would result in muscle ischemia, injury and fatigue during exercise.
We previously reported that the blockade of NRG signaling through ERBB2/4 receptors prevented phosphorylation of α-dystrobrevin1 and hence affected NMJ stability1. However ablation of ERBB2/4 receptors, and thus elimination of NRG signaling through them, results in a lack of dystrophin expression (Figure 1). Thus NRG/ERBB signaling maintains NMJ stability through at least two pathways, one where it phosphorylates α-dystrobrevin 11,2 and the other where it stimulates dystrophin expression and thereby allowing the formation of a DAPC that stabilizes acetylcholine receptors (Figure 2).
NRG/ErbB signaling also induces cardiomyocyte proliferation and repairs heart injury12 and is essential for normal cardiac development9–11, whereas dystrophin deficiency does not impair cardiac development but does result in dilated cardiomyopathy (DCM)8. Since ERBB4 can heterodimerize with ERBB2 and a function blocking ERBB2 antibody treatment results in DCM in mice21 and DCM in cancer patients with a function blocking HER2 antibody treatment25, this is consistent with NRG/ERBB signaling stimulating dystrophin expression, since DMD patients and mdx mice, both lacking functional dystrophin, develop DCM (Figure 2). Hence the data presented here provides a mechanism for the reported beneficial effects of ERBB4 in repairing heart injury whereby NRG/ERBB2/4 signaling stimulates dystrophin expression. Thus NRG/ERBB carries out different functions in cardiac development and maintenance through different signaling targets, in the latter case through regulating dystrophin expression.
Increasing NRG signaling through ERBB2/4, especially in the smooth muscle of blood vessels, could be a way to increase truncated dystrophin expression in D/BMD patients. This would ameliorate dystrophic symptoms in those patients where the mutation in dystrophin does not affect association of nNOS26 and thereby enabling normal blood flow during exercise. Furthermore, as dystrophin is also present in all regions of the brain, being most abundant in the cerebellum27, and since NRG/ERBB signaling regulates dystrophin expression, levels of dystrophin could be the underlying cause of some of the disease states such as schizophrenia, associated with NRG/ERBB function in the brain.
NRG signaling through ERBB2/4 receptors is necessary for stimulation of dystrophin expression. However, when ERBB2/4 receptors are lacking in skeletal muscle but expressed in smooth muscle, mice do not exhibit dystrophic symptoms demonstrating that lack of dystrophin expression in smooth muscle is the root cause of the onset of D/BMD.
Dataset 1. Uncropped western blot images and raw Ct values from qPCR. DOI: https://doi/org/10.5256/f1000research.15889.d21462828.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)