Keywords
S6K1, subcellular localization, mTOR/S6K1 signaling pathway, breast cancer, TBR2, Eomesodermin, MCF-7 cell line
S6K1, subcellular localization, mTOR/S6K1 signaling pathway, breast cancer, TBR2, Eomesodermin, MCF-7 cell line
Ribosomal protein S6 kinase 1 (S6K1) belongs to the AGC family of serine/threonine protein kinases (Ruvinsky & Meyuhas, 2006). It is involved in the regulation of crucial physiological processes, such as protein synthesis, ribosomal biogenesis, the G1/S-phase transition of the cell cycle, mRNA splicing, differentiation of specific cell types, and apoptosis. A large number of cellular targets makes S6K1 a key regulator of cell size, growth, and proliferation (Magnuson et al., 2012). S6K1 activity is under the control of the PI3K/Akt/mTOR signaling pathway, which is dysregulated in diverse human pathologies, including diabetes, obesity, neurodegenerative disorders, and cancer (Tavares et al., 2015). Overexpression of S6K1 was found in several tumor types, including breast cancer, and this was associated with a worse disease outcome (Bostner et al., 2015).
In mammalian cells, S6K1 is encoded by RPS6KB1 gene located at chromosome 17. Several isoforms of the S6K1 protein are known: 85kDa S6K1 and 70kDa S6K1 (p85S6K1 and p70S6K1 respectively), which originate from alternative translation initiation sites, and hypothetical p60S6K1, suggested to be a product of alternate mRNA translation as well (Kim et al., 2009). Recently, new 31kDa isoform of S6K1 (p31S6K1) encoded by mRNA splice variant was identified. It was demonstrated that translated p31S6K1 isoform does not have catalytic activity, but possesses oncogenic properties (Ben-Hur et al., 2013; Song & Richard, 2015). The longer isoform p85S6K1 has an additional 23 amino acid extension at the N-terminus of the molecule where the nuclear localization signal is located. Earlier p85S6K1 was described as a predominantly nuclear kinase. However, recent studies revealed it in the cytoplasm of the breast cancer cells and in the primary human fibroblasts using nuclear-cytoplasmic fractionation (Kim et al., 2009; Rosner & Hengstschläger, 2011). Another isoform of S6 kinase p70S6K1 was thought to localize predominantly in the cytoplasm. Treatment of cells with leptomycin B (the nuclear export inhibitor) led to the accumulation of the p70S6K1 in the nucleus. This finding allowed supposition that p70S6K1 shuttles between the cytoplasm and nucleus of the cell (Panasyuk et al., 2006). To date, very little is known about p31S6K1 subcellular localization. It is thought to be nuclear in human normal fibroblasts (Rosner & Hengstschläger, 2011). Overall, S6K1 subcellular localization data have been based predominantly on subcellular fractionation assay or immunocytochemical analysis of recombinantly expressed kinase. Information about nucleocytoplasmic distribution of the endogenous S6K1 is still limited, and mechanisms of its regulation remain elusive.
Recent studies suggest that S6K1 subcellular localization and activation depends on physiological features of different tissues. Immunohistochemical analysis of breast tumors revealed prominent S6K accumulation in the nuclei of carcinoma cells (Filonenko, 2013; Filonenko et al., 2004; Lyzogubov et al., 2005). In other studies, it was shown that nuclear accumulation of S6K1 was indicative of a reduced tamoxifen effect in breast cancer patients, while cytoplasmic localization of S6K1 was associated with better prognosis (Bostner et al., 2015).
Migration of the cancer cells is an important stage of cancer progression that usually lead to the tissue invasion and formation of distant metastases. The recent data suggest that S6K1 could be involved in the regulation of the motility of normal and malignant cells. Knockdown of p70S6K1 or inhibition of S6K1 kinase activity causes a significant decrease in the migration properties of the prostate, breast, and ovarian cancer cells in vitro (Amaral et al., 2016; Ip et al., 2011). Moreover, activation of p70S6K1 in human ovarian carcinoma cells that occurs in response to stimulation by hepatocyte growth factor (HGF) led to increased expression of matrix metalloproteinase 9 (MMP9) and higher migration rate of these cells (Zhou & Wong, 2006). It was shown that p70S6K1 stimulated activation of Cdc42, Rac1, and PAK1 – the known regulators of actin cytoskeleton reorganization (Aslan et al., 2011; Liu et al., 2010). Besides, S6K1 colocalizes with the actin arches at the leading edge of moving mesothelioma cells. Treatment with rapamycin (specific mTOR inhibitor) complicated the formation of actin arches even when cells were stimulated with endothelial growth factor (EGF) (Berven et al., 2004; Liu et al., 2008). However, the link between subcellular localization of S6K1 and its functions in migrating cancer cells is not fully understood.
In the present research, we studied the subcellular localization of endogenous S6K1 in breast tumor and normal tissue, and in breast adenocarcinoma MCF-7 cells in monolayer culture, 3D multicellular spheroids, and in course of cancer cell migration. We found that nucleocytoplasmic distribution of S6K1 depends greatly on the density of the monolayer culture, and is different in 3D vs 2D cell culture. Moreover, we revealed that S6K1 relocalizes to the nucleus during migration of cancer cells from multicellular spheroids onto growth surface. In addition, we analyzed the possible interaction of S6K1 with a number of transcription factors, involved in the regulation of cell motility. For the first time, we described the colocalization and co-immunoprecipitation of S6K1 and TBR2 (T-box brain protein 2) in breast cancer cells. These data could indicate that during cell migration S6K1 possibly interacts with transcription factors in the cell nucleus, activating genes that are involved in the regulation of cell locomotor activity.
Human breast adenocarcinoma cell line MCF-7 was obtained from Bank of Cell Lines of the R. E. Kavetsky Institute of Experimental Pathology, Oncology and Radiobiology, NASU (Ukraine). The cells were cultivated in DMEM culture medium (Gibco, USA) supplemented with 10% fetal calf serum (FCS, HyClone, USA), 4 mM glutamine, 50 units/ml penicillin, 50 µg/ml streptomycin at 37°C under 5% CO2. The medium was changed every third day. For immunofluorescence analysis cells were seeded onto sterile glass coverslips.
To form multicellular spheroids, MCF-7 cells were detached and 1×106 cells were seeded into 100 mm Petri dishes coated with 1% agarose (Sigma-Aldrich, A9045) for 72 h.
For initiation of spheroid-to-monolayer reversion and cell migration, multicellular spheroids were transferred onto growth surface (glass coverslip) and cultivated for 24 or 72 h. Then outspreaded spheroids were applied to immunofluorescence analysis.
Cellular and spheroid morphology was evaluated microscopically using transmitted light (CETI Versus inverted microscope, CETI, Belgium, and Leica DM 1000, Leica Microsystems, Germany).
Cultured MCF-7 cells were fixed with 10% formalin for 15 min at room temperature (RT). Thereafter, the cells were permeabilized with 0.2% Triton X-100 in PBS for 10 min. Autofluorescence was decreased by incubation with 10 mM cupric sulphate and 50 mM ammonium acetate, pH 5.0 for 30 min at RT. Non-specific binding was blocked after incubation with 10% FCS in PBS for 30 min at 37°C in a humidifying chamber.
S6K1 subcellular localization was revealed using anti-S6K1-C-terminus rabbit polyclonal antibodies (generated and evaluated earlier (Savinska et al., 2001; if you are interested in obtaining this antibody, please contact the corresponding author)) at 1:100, and anti-phospho-S6K1 (T389) rabbit polyclonal antibodies at 1:20 (Cell Signaling Technology Cat# 9205, RRID:AB_330944). The secondary Fluorescein (FITC)-AffiniPure Goat Anti-Rabbit IgG (H+L) antibody 1:400 (Jackson ImmunoResearch Labs Cat# 111-095-003, RRID:AB_2337972) were applied for 45 min at 37°C in a humidifying chamber.
Double immunofluorescence analysis was performed by addition of the primary antibody mix: anti-S6K1-C-terminal mouse monoclonal antibodies (generated earlier (Pogrebnoy et al., 1999); if you are interested in obtaining this antibody, please contact the corresponding author) at 1:100 + anti-TBR2 rabbit polyclonal antibodies at 1:100 (Abcam Cat# ab23345, RRID:AB_778267), or + anti-ERG rabbit monoclonal antibodies at 1:50 (Dako, Cat#M7314), or anti-CDX2 rabbit monoclonal antibodies at 1:100 (Abcam Cat# ab76541, RRID:AB_1523334), overnight at +4°C in a humidifying chamber. The secondary Fluorescein (FITC)-AffiniPure Donkey Anti Mouse IgG (H+L) antibody (Jackson ImmunoResearch Labs Cat# 715-095-150, RRID:AB_2340792) at 1:400, and Rhodamine (TRITC)-AffiniPure Donkey Anti-Rabbit IgG (H+L) antibody (Jackson ImmunoResearch Labs Cat# 711-025-152, RRID:AB_2340588) at 1:400 were applied for 45 min at 37°C in a humidifying chamber. Samples were embedded into Mowiol medium (Sigma-Aldrich, USA) containing 2.5% DABCO (Sigma-Aldrich), 0.5 % DAPI (Sigma-Aldrich).
All microscopy studies were performed using Leica DM 1000 fluorescent microscope and Zeiss LSM 510 META microscope (Carl Zeiss Microscopy GmbH, Germany). Fluorescence images were analyzed with free software Fiji/ImageJ v1.52b (Fiji, RRID:SCR_002285; Schindelin et al., 2012). Figures were generated with the FigureJ plugin (Mutterer & Zinck, 2013) in Fiji/ImageJ v1.52b.
For quantitative characterization of colocalization Pearson coefficient and Manders coefficients (M1 and M2) analysis was performed on background-subtracted images using JACoP plugin (Bolte & Cordelières, 2006) in Fiji/ImageJ v1.52b. Pearson coefficient (Rr) and Manders coefficients (M1 and M2) were expressed as mean value +/-SD, the experiment was duplicated, n=5 for each condition. To validate and describe the obtained degree of colocalization pre-defined image sets from Colocalization Benchmark Source were used. Obtained values of the colocalization coefficients were used to find the closest benchmark.
Histological samples of human mixed ductal/lobular carcinoma of the breast and surrounding conditionally normal tissue were obtained from 10 patients within the framework of the cooperation agreement between the National Cancer Institute and the Institute of Molecular Biology and Genetics of the National Academy of Sciences of Ukraine. This study has been approved by the Committee on Biological & Medical Ethics of the National Cancer Institute of Ukraine (approval number - № 67, 25.03.2015). Written informed consent was obtained from all patients for the use of their tissues in research.
Sections of human breast cancer and surrounding tissues or multicellular spheroids were deparaffinized in xylene and rehydrated in a series of graded alcohols. For the antigen retrieval, slides were placed in citrate buffer (10 mM citric acid, pH 6.0) and transferred to the microwave. Sections were boiled two times for 5–7 min. Then, sections were treated with 0.2% Triton X-100 for 10min. Endogenous peroxidase was quenched with 3% H2O2 in PBS for 30 min. After blocking of non-specific staining with 10% FCS in PBS, sections were incubated with anti-S6K1-C-terminal rabbit polyclonal antibodies (1:100) overnight at +4°C, and thereafter they were incubated with peroxidase-conjugated secondary antibodies (1:100; Promega Cat# W4011, RRID:AB_430833) for 1 hour at 37°C. The reaction was developed with 3,3’diaminobenzidine (Sigma-Aldrich) solution.
Predicted sites of TBR2 phosphorylation by S6K1 were revealed using Group-based Prediction System v2.1 (Xue et al., 2011; GPS, RRID:SCR_016374). The sequence of human TBR2 for this analysis was taken from the National Center for Biotechnology Information, NCBI Reference Sequence: NP_001265111.1.
Anti-S6K1 mouse monoclonal antibodies (Pogrebnoy et al., 1999) were immobilized on protein A/G PLUS Agarose beads (Santa Cruz Biotechnology) overnight at +4 C.
MCF-7 cells were washed with ice-cold phosphate-buffered saline and extracted with lysis buffer, containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100, 5mM EDTA, 50 mM sodium fluoride, 5 mM β-glycerophosphate,10 mM sodium pyrophosphate, 1 mM sodium orthovanadate and a mixture of protease inhibitors (Roche Molecular Diagnostics, France). Cell lysates were centrifuged at 13 000 rpm for 20 min at 4°C. Endogenous S6K1 was precipitated by adding 1000µg of total cell lysates to the immobilized antibodies and incubating overnight at 4°C. Immune complexes were washed three times with lysis buffer, boiled for 5 min in Laemmli sample buffer, and used for immunoblot analysis. As a control, protein A/G PLUS Agarose beads were incubated with monoclonal antibodies or cell lysates alone.
Samples were separated by 10% SDS-PAGE and transferred onto PVDF membrane (Millipore, Billerica, MA). The membrane was blocked with 5% skim milk in PBST (140 mM NaCl, 2.6 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, 0,05% Tween-20, pH 7.4) for 1 h at RT, and then incubated with anti-TBR2/Eomes rabbit antibodies at 1:500 (Abcam Cat# ab23345, RRID:AB_778267) or anti-S6K1 C-terminal rabbit polyclonal antibodies 1:3000 overnight at 4°C. After washing three times with PBST, HRP-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch Labs Cat# 111-035-144, RRID:AB_2307391) in 1:10 000 dilution were incubated with the membrane for 1 h at RT. Finally, the membrane was developed using an enhanced chemiluminescence system (Fluco) and then exposed to Agfa X-ray film.
There are several mediated and direct approaches to determine protein function in cells. The first includes cell models without direct interaction between the studied protein and the reporter marker. The function of a studied protein is evaluated as a correlation with a reporter marker, which in turn gives a characteristic to a particular cellular process (Nizheradze, 2006). The second group of approaches is based on closer interactions between the agent and the reporter marker. One of them consists of the determination of studied protein belonging to the specific subcellular compartment and next revealing its protein-partners with known functions. The difference in subcellular localization of the protein in different cell types may suggest the realization of different function.
For the first step of the present study, subcellular distribution of S6K1 was analyzed in normal and cancer cells of a histological section of human breast cancer and normal tissues. Our results support previous data about preferential nuclear localization of S6K1 in breast malignant cells (Bostner et al., 2015; Filonenko et al., 2004) and mainly cytoplasmic one in conditionally normal surrounding tissues (Figure 1A, B).
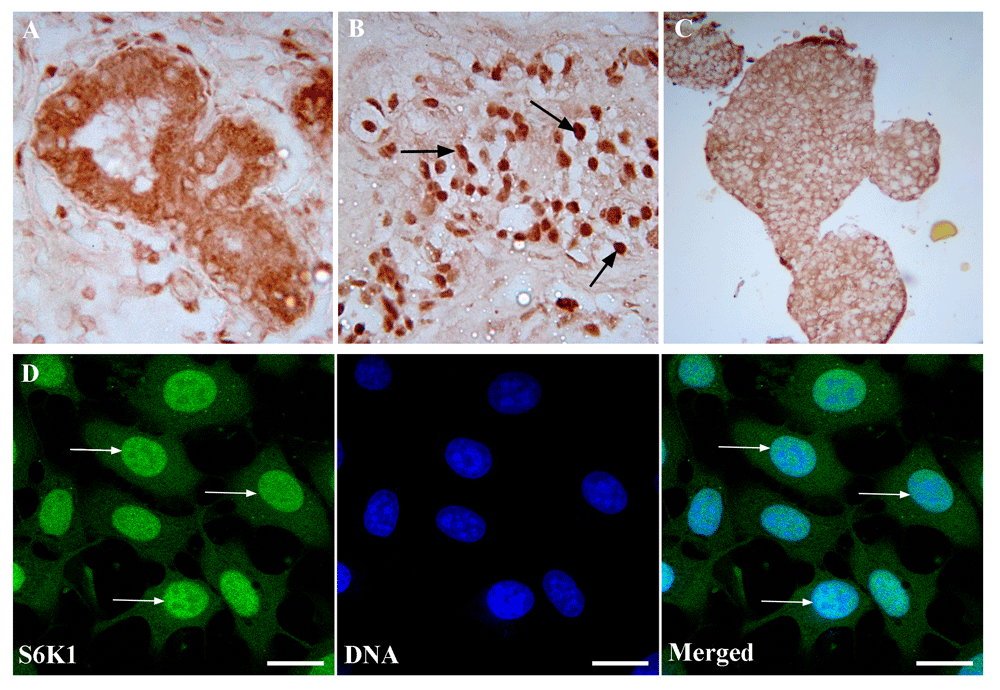
(A) Immunohistochemical detection of S6K1 in human conditionally normal breast tissue. Magnification 400x. (B) Immunohistochemical analysis of S6K1 subcellular localization in human breast cancer tissue. Magnification 400x. Arrows point out to the positive reaction in the nuclei of the cells. (C) Immunohistochemical detection of S6K1 in MCF-7 multicellular spheroids. Magnification 200x. (D) Immunofluorescence image of S6K1 subcellular distribution (green) in 40% confluent MCF-7 cell monolayer. Arrows point out to the positive reaction in the nuclei of the cells. DNA was counterstained with DAPI (blue). Scale bars correspond to 20 µm. The data are representative of three independent experiments.
As was shown earlier, 3D cell culture systems are preferable for tumor growth study (Bingel et al., 2017). That’s why we analyzed the intracellular distribution of S6K1 in multicellular spheroids of MCF-7 cells and in monolayer culture. We detected a bright signal of S6K1 in the cytoplasm and its absence in the nuclei of cells of the multicellular spheroids (Figure 1C). In contrast to a spheroid culture, S6K1 distribution in MCF-7 cells monolayer culture at 40–60% confluence revealed bright nuclear staining in interphase cells with the moderate cytoplasmic signal (Figure 1D).
A significant difference in S6K1 localization in monolayer and spheroid cultures can be explained by differences in cell growth conditions in two distinct cultures. First of all, the reason for such alterations could be explained by a cascade of intracellular events induced by cell-matrix adhesion or intercellular interactions. One can assume that the S6K1 is involved in such intracellular rearrangement. To clarify this issue, S6K1 subcellular localization in MCF-7 cells cultured at different densities was analyzed by the present study. The immunofluorescence analysis revealed the displacement of S6K1 from the nucleus to the cytoplasm when the cell culture density increased (Figure 2A–E). At the lowest MCF-7 cell density, S6K1 was observed predominantly in the nuclei of cultured cells whereas at the highest density the main signal was concentrated in the cytoplasm.
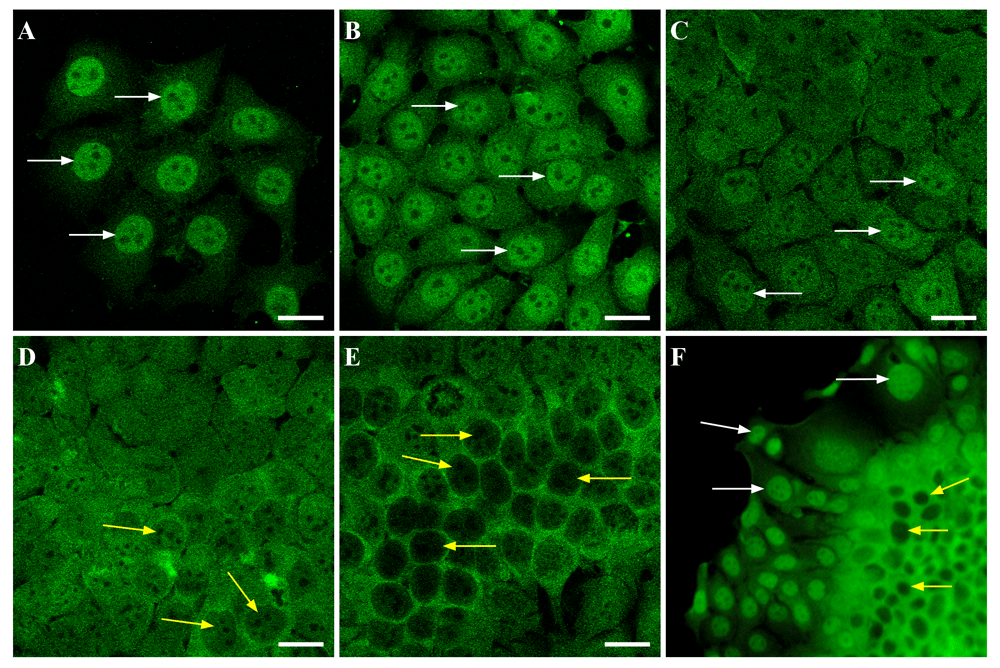
MCF-7 cells were seeded onto glass coverslips in the density 10 000 cells/well (A), 30 000 cells/well (B), 50 000 cells/well (C), 70 000 cells/well (D), 100 000 cells/well (E), and cultivated for 48 hours. Then cells were fixed and stained with anti-S6K1 (green). White arrows point to the prominent positive reaction in the nuclei of the cells, yellow arrows point to the decreased staining in the nuclei. Scale bars are 20 µm. The images are representative of three independent experiments. (F) MCF-7 cells were cultivated to form a super-confluent monolayer. Then fragments of the monolayer were gently detached by short incubation with trypsin, transferred on the coverslip and left for 48 hours to grow. After it the fragments were fixed and stained with anti-S6K1 (green). White arrows point to the positive reaction in the nuclei of the cells at the leading edge of the monolayer fragment; yellow arrows point to the decreased staining in the nuclei of the cells at the center of the monolayer fragment. Magnification 400x.
Besides, to test the possible dependence of S6K1 subcellular localization on cell density the following approach was introduced. After reaching 90% confluence layer of MCF-7 cells, the monolayer was detached from growth surface by short treatment with trypsin (w/o EDTA), and replaced in fresh culture medium. After cultivation for 48 h such fragments of monolayer demonstrated high cell density in the center and spread out cells at the edges. Immunofluorescence analysis revealed that cells’ spreading was accompanied by the alterations in S6K1 localization (Figure 2F). Densely packed cells in the center of the fragments demonstrated cytoplasmic reaction for S6K1, while spread out cells had preferentially nuclear staining.
Since cell spreading can be considered as a stage of migration, the previous data led to the hypothesis of a relation between the initiation of cell migration and the relocalization of S6K1. Among the variety of cell migration models, it is necessary to point out those that are based on 3D cell cultures (Metzger et al., 2017). There are a lot of data concerning the similarity of multicellular spheroid organization and structure of solid malignant tissue (Rodrigues et al., 2018). Besides, the transformation of 3D multicellular spheroids in 2D cell colonies can be realized only by activation of cultured cell migration. So, in the present study, S6K1 distribution was examined on migrating MCF-7 cells after replacing MCF-7 multicellular spheroids onto the growth surface (Figure 3). We applied the immunofluorescence analysis of cultured cells after 24 and 72 hours after initiation of cell migration from MCF-7 spheroids. Obtained data suggest that significant relocalization of S6K1 from the cytoplasm into the nuclei in course of locomotor function realization took place (Figure 4A, B). The cells, which were still in 3D condition, had positive cytoplasm and negative nuclei, as well as cells of spheroid at histological sections regardless of their remoteness from the edge of the spheroid (Figure 1C). During MCF-7 spheroid transformation in monolayer, spreading cells demonstrated strong accumulation of S6K1 in the nuclei (Figure 4B).
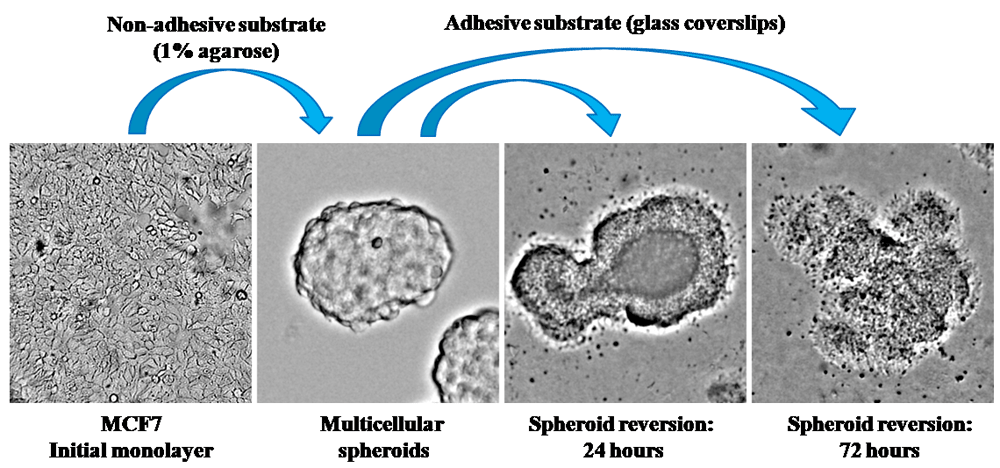
To generate multicellular spheroids, MCF-7 cells were seeded in the Petri dishes coated with 1% agarose, and cultivated for 72 hours. To analyze cell migration, obtained spheroids were transferred onto glass coverslips and cultured for 24 or 72 hours. Microphotographs of the spheroids general view were taken by Leica DM1000 (Leica, Germany) in transmitted light. Magnification 200x.
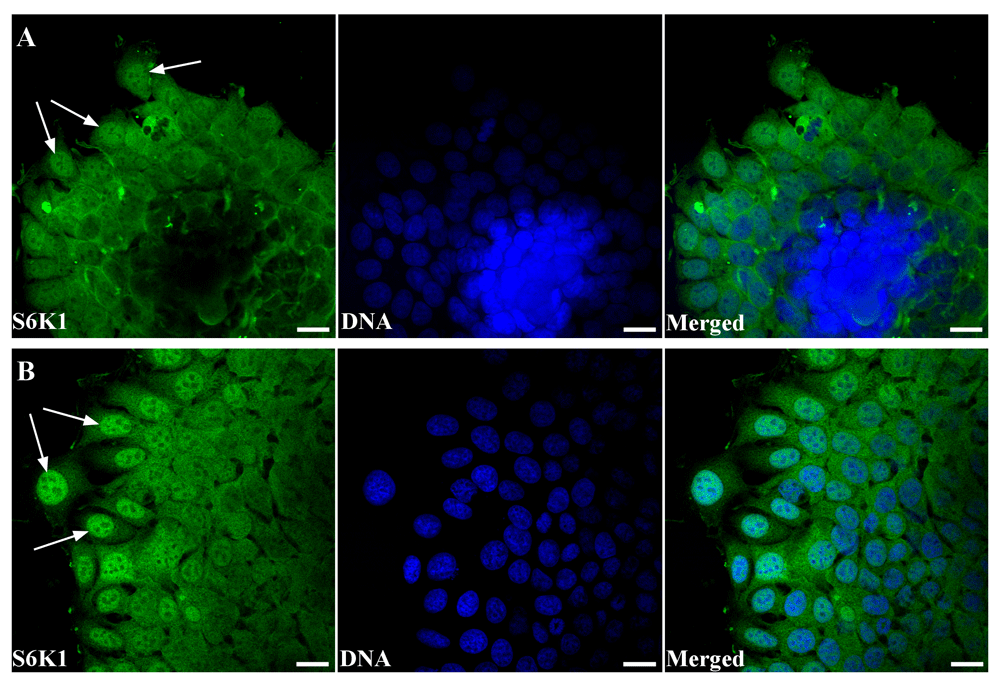
(A) Immunofluorescence analysis of S6K1 subcellular localization (green) in the MCF-7 spheroid reversed for 24 hours. Arrows point to the predominantly nuclear distribution of the S6K1 in the migrating cells. DNA was counterstained with DAPI (blue). Scale bars are 20 µm. (B) Immunofluorescence analysis of S6K1 subcellular localization (green) in the MCF-7 cells at the leading age of spheroid reversed for 72 hours. Arrows point to the predominantly nuclear distribution of the S6K1 in the migrating cells. DNA was counterstained with DAPI (blue). Scale bars are 20 µm. The images are representative of three independent experiments.
The most often used marker of S6K1 activation is Thr389 phosphorylation mediated by mTOR (Romanelli et al., 2002). Thus, we analyzed phosphorylation status of S6K1 in MCF-7 cells during spheroid transformation into monolayer by immunofluorescence analysis. Overall, the pattern of phospho-S6K1 distribution was similar to that observed for total S6K1 (Figure 5). Namely, in the central part of the spheroid, S6K1 was mainly observed in the cytoplasm (however some of the nuclei were positive), whereas all cells of the leading edge demonstrated predominant nuclear kinase localization (Figure 5A). Also, strong nuclear localization of phospho-S6K1 (Thr389) was revealed in monolayer culture of MCF-7 cells (Figure 5B).
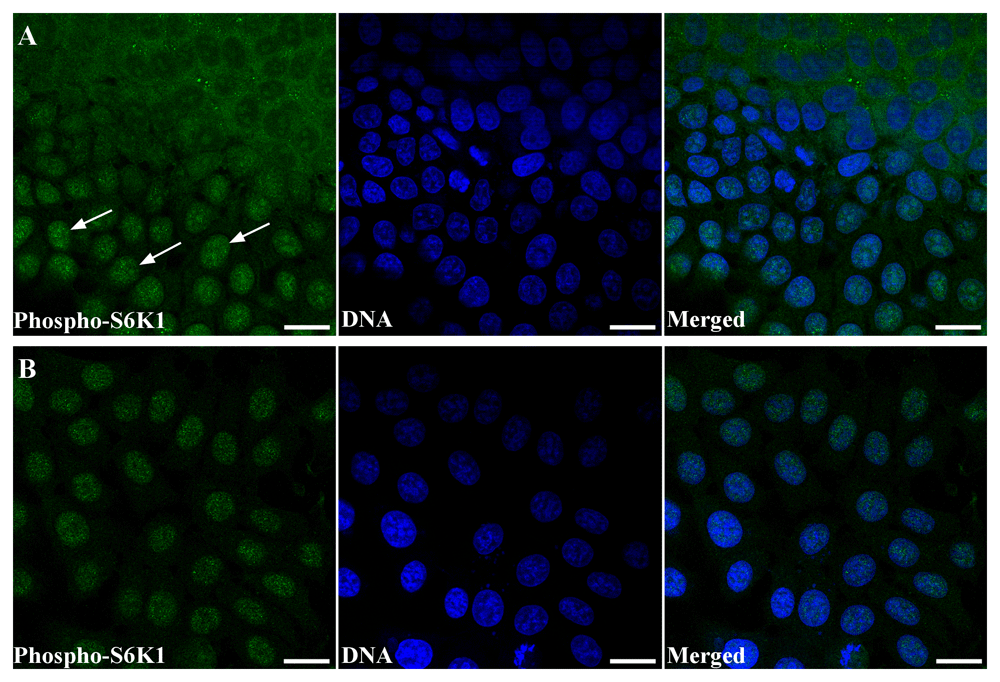
(A) Confocal image of MCF-7 spheroid-to-monolayer reversion model. Cells were stained with anti-phospho-S6K1 (T389) (green). DNA was counterstained with DAPI (blue). Scale bars correspond to 20 µm. Arrows point to the positive reaction in the nuclei of the migrating cells at the leading edge of the spheroid. (B) Confocal image of monolayer culture of the MCF-7 cells stained with anti-phospho-S6K1 (T389) (green). DNA was counterstained with DAPI (blue). Scale bars correspond to 20 µm. The images are representative of two independent experiments.
Obtained data suggested that activation of cell locomotor function is accompanied by cytoplasm/nuclear shuttling of S6K1; however, the biological sense of the event is not yet clear. The possible explanation could be the implication of S6K1 in the regulation of transcription factors affecting expression of genes that control cell migration.
As it was mentioned above, several transcription factors are known as targets of S6K, but their nuclear localization in relation to cell migration was not reported. That’s why, we analyzed subcellular distribution of several transcription factors, which are mTOR/S6K signaling regulated and activated in migrating cells either in a cancer tissue or in the process of organism development. Among them the mammalian transcription factor CDX2, which plays a key role in intestinal development and differentiation. It was determined that reduced expression of CDX2 is important in colon tumorigenesis through mTOR-mediated chromosomal instability (Aoki et al., 2003). Fusion of another transcription factor ERG and androgen-responsive TMPRSS2 serine protease is an important feature of prostate cancer. A strong correlation has been revealed between TMPRSS2-ERG fusion and activation of mTOR/S6K pathway (Faraj et al., 2013; King et al., 2009). The third studied transcription factor was T-box transcription activator Eomesodermin (or TBR2) (Conlon et al., 2001). Earlier it was regarded as a target for anticancer therapy. It was detected that siRNA knockdown of Eomesodermin in human hepatocellular carcinoma significantly affected anchorage-independent cell growth (Gao et al., 2014). Besides, it is involved in lymphocyte differentiation. It should be noted that TOR signaling is involved in modulation of TBR2 activity; it was demonstrated that TOR signaling was the central regulator of transcriptional programs by regulation of expression of transcription factors T-bet and Eomesodermin, that determined effector or memory cell fates in CD8+ T cells (Cui et al., 2016).
Immunofluorescence analysis of subcellular distribution of S6K1 and mentioned transcription factors in MCF-7 cells revealed that ERG transcription factor either was present in scant quantities or not determined at all in MCF-7 cells (Dataset 4; (Kosach et al., 2018d)). CDX2 was determined as positive dots predominantly in the nuclei, CDX-2 and S6K1 colocalization was not detectable by confocal microscopy (Dataset 4; (Kosach et al., 2018d)). TBR2/Eomesodermin positive granules were observed in the cytoplasm as well as in nuclei (Figure 6A, B). In both cases, partial but bright colocalization of TBR2 and S6K1 was revealed. Moreover, in low-density monolayer, when S6K1 localized mainly in the nuclei, TBR2 was observed predominantly in the nuclei as well. At high-density monolayer, S6K1 was redistributed in the cytoplasm, and TBR2 repeated the pattern of immunofluorescent reaction (Figure 6A, B).
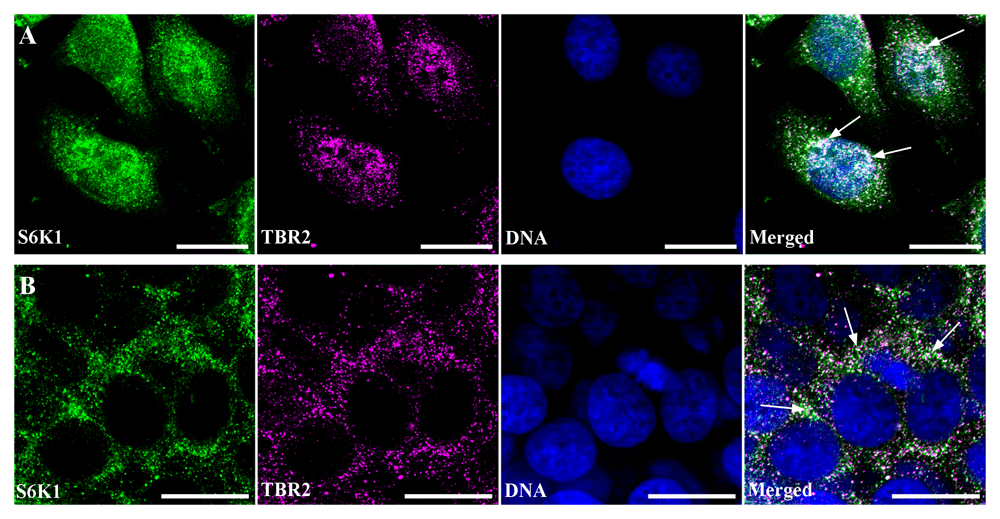
(A) Immunofluorescence image of low density monolayer culture of the MCF-7 cells double stained with anti-S6K1 (green) and anti-TBR2 (magenta). DNA was counterstained with DAPI (blue). Scale bars correspond to 20 µm. Arrows point to the regions of S6K1 and TBR2 colocalization. (B) Immunofluorescence image of high density monolayer culture of the MCF-7 cells double stained with anti-S6K1 (green) and anti-TBR2 (magenta). DNA was counterstained with DAPI (blue). Scale bars correspond to 20 µm. Arrows point out to the regions of S6K1 and TBR2 colocalization. The images are representative of two independent experiments.
For quantitative characterization of S6K1 and TBR2 colocalization, Pearson coefficient (Rr) and Manders coefficient (M1 and M2) analysis was performed on background-subtracted images using JACoP ImageJ plugin (Bolte & Cordelières, 2006). M1 shown the colocalization of S6K1 with TBR2, whereas M2 expressed the pool of TBR2 colocalizing with S6K1. Colocalization analysis of S6K1 and TBR2 in low density monolayer revealed Pearson coefficient Rr= 0.55 +/- 0.113, M1= 0.999 +/-0.01, M2= 0.84 +/-0.087. To validate and describe the obtained degree of colocalization pre-defined image sets from Colocalization Benchmark Source were used. The closest benchmark was CBS007RGM that corresponded to 60% colocalization, thus indicating the medium level of colocalization between TBR2 and S6K1. A slightly lower but reliable colocalization of S6K1 and TBR2 was observed in a monolayer with a high density. Namely Pearson coefficient was Rr=0.47 +/- 0.064, Manders coefficients were M1=0.995 +/-0.004 and M2=0.62+/-0.187. So, a slightly higher level of S6K1 and TBR2 colocalization was revealed in MCF-7 cells grown in low density monolayer, when S6K1 and TBR2 localized mainly in the nuclei.
The application of immunoprecipitation confirmed the interaction of S6K1 and TBR2 (Figure 7). Protein complexes containing S6K1 were extracted from cultured MCF-7 cell lysate using anti-S6K1 antibodies and then blotted with antibodies to TBR2. Obtained results revealed protein complex formation of S6K1 and TBR2 suggesting the possibility of TBR2 regulation via S6K1 mediated phosphorylation.
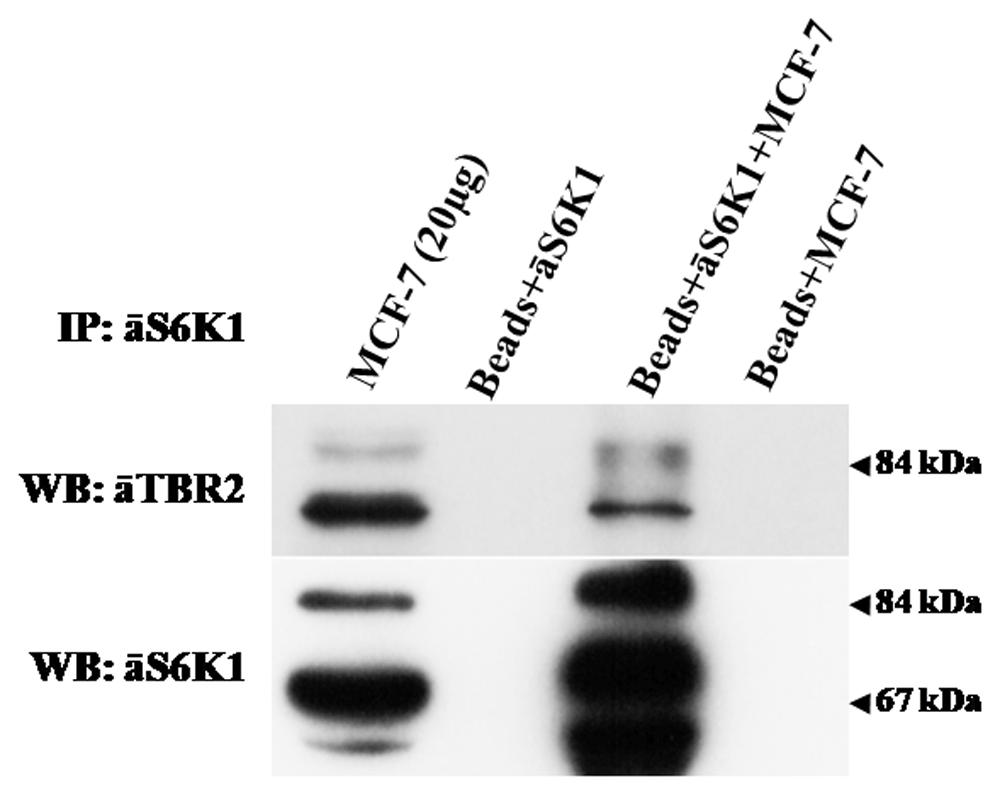
Endogenous S6K1 was precipitated with anti-S6K1 mouse monoclonal antibodies immobilized on protein A/G PLUS Agarose beads (Santa Cruz Biotechnology). As a control, protein agarose beads were incubated with monoclonal antibodies or cell lysates alone. Immune complexes were analyzed by immunoblotting with anti-TBR2 rabbit antibodies (Abcam, ab23345) or anti-S6K1 C-terminal rabbit polyclonal antibodies. The data are representative of two independent experiments.
Further computational prediction of phosphorylation sites in TBR2 (GPS 2.1) indeed revealed several sites, and three of them (Tyr421, Tyr423, Ser646) can be phosphorylated by S6K1 with a high score (Figure 8). Interestingly, both Tyr421 and Tyr423 are located in the DNA binding domain and one can assume that their phosphorylation could be related to the binding ability of the transcription factor to the targeted DNA. Another phosphorylation site (Ser646) is located within transcription activation domain at C-terminus of TBR2, which is involved in transcription activation. So, S6K1 can be involved in the regulation of TBR2 transcription activity. However, further research is needed to find if S6K1 phosphorylates TBR2 in vitro and in vivo.
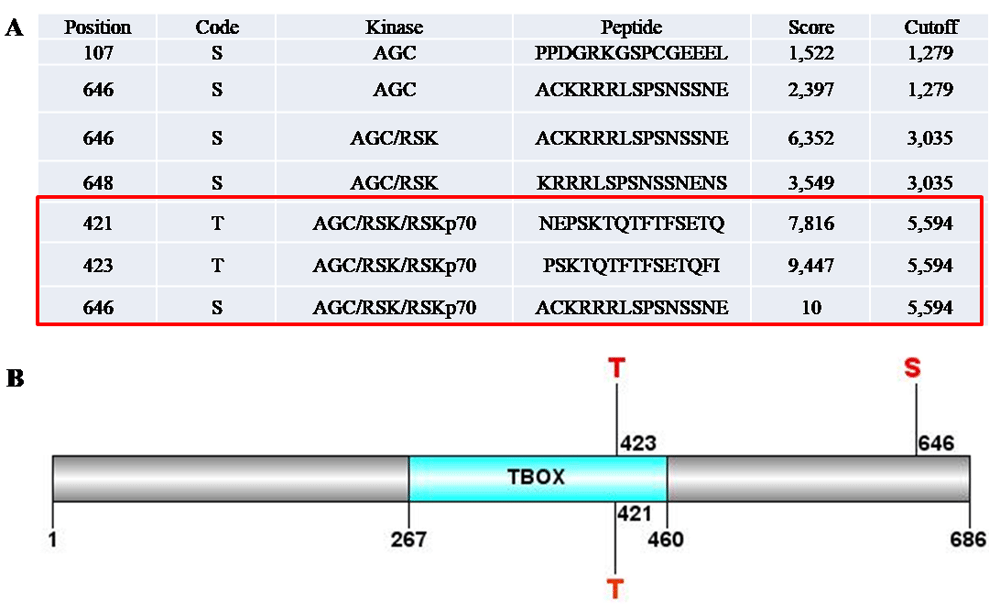
Free software Group-based Prediction System v2.1 was used for bioinformatics analysis. It revealed that TBR2 contained three sites that could be phosphorylated by S6K1 with a high probability (A). Two of them, T421 and T423, situates in the DNA binding domain of the TBR2. Third site S646 is within transcription activation domain at C-terminus of TBR2 (B). Possible phosphorylation of these residues by S6K1 could significantly influence the TBR2 activity. However, this finding warrants further research.
Concerning TBR2 targets, in the course of organism development, Eomesodermin can induce virtually the entire spectrum of mesodermal genes in all types of mesodermal cells, which could appear in malignant cells of non-mesodermal origin (Reim et al., 2017; Russ et al., 2000).
Considering the multiplicity of S6K1 substrates, phosphorylation of the TBR2 transcription factor is not the only reason for the movement of the kinase from the cytoplasm into the nucleus of migrating cells. However, the proposed interaction can partially explain the accumulation of kinase in the nucleus of moving cells. The existence of several kinase isoforms may explain the partial colocalization of the kinase and the transcription factor (Amaral et al., 2016). In addition to the previously known classical nuclear substrates of S6K1, in case of breast cancer, it is necessary to note that this kinase can activate estrogen receptor-α, which is a nuclear transcription factor by its phosphorylation at Ser167 in a ligand-independent manner (Yamnik & Holz, 2010). Besides, recent data indicate that S6K1 is targeted by histone acetyltransferases p300 and p300/CBP-associated factor (PCAF). The significance of this acetylation is not fully clear, but by analogy with S6K2, it is assumed that S6K1 is involved in the regulation of the transcription process (Fenton et al., 2010). Summing up, there are a number of data confirming the nuclear localization of S6K1, but the role that S6K1 performs in the nucleus of migrating malignant cells require further investigation.
For the first time, this study revealed S6K1 relocalization from the cytoplasm to the nuclei in migrating cells using the model of spheroids MCF-7 cells transformation into monolayer culture. Such relocalization could be linked to the S6K1 driven activation of transcription factors responsible for cell locomotion. Particularly, colocalization and interaction of S6K1 and transcription factor TBR2 were revealed using confocal microscopy and co-immunoprecipitation. In addition, bioinformatics analysis of phosphorylation sites in TBR2 supports a prediction about S6K1 mediated phosphorylation and regulation of TBR2.
F1000Research: Dataset 1. Unedited images that were used in Figure 1 and Figure 2, showing S6K1 subcellular localization in breast normal tissue, cancer tissue, and in MCF-7 cells monolayer., 10.5256/f1000research.15447.d214430 (Kosach et al., 2018a).
F1000Research: Dataset 2. Unedited images from Figure 3., 10.5256/f1000research.15447.d214431 (Kosach et al., 2018b).
F1000Research: Dataset 3. Unedited images that were used in Figure 4 and Figure 5, showing S6K1 and phospho-S6K1 (T389) subcellular localization during MCF-7 cell migration., 10.5256/f1000research.15447.d214432 (Kosach et al., 2018c).
F1000Research: Dataset 4. Unedited images of S6K1 colocalization with transcription factors TBR2 (Figure 6), ERG, and CDX2., 10.5256/f1000research.15447.d214433 (Kosach et al., 2018d).
F1000Research: Dataset 5. Unedited western blot images of co-immunoprecipitation of S6K1 and TBR2 used in Figure 7., 10.5256/f1000research.15447.d214434 (Kosach et al., 2018e).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)