Keywords
Differential gene expression analysis, pathway analysis, combining p-value, cell signalling network
Differential gene expression analysis, pathway analysis, combining p-value, cell signalling network
High throughput sequencing techniques have been widely used to yield differentially expressed genes (DEG) (Malone & Oliver, 2011). To this end, changes in transcript abundance are measured, e.g. by next generation sequencing techniques, and interpreted as an indicator of differential expression of genes. DEGs can be used to gain insights into the mechanism underlying differences between conditions of samples, such as healthy versus diseased. Differential gene expression analysis (DGEA) informs about the magnitude of expression changes between the conditions which are often expressed as fold change, sign of fold change and the confidence level of observing an authentic change, often expressed as p-value. These DEGs information is further interpreted to extract meaningful biological insights. For example, genes that could be involved in the response to a particular stimuli or maybe the cause of a disease. To this end, pathway-based analysis has become an important tool to further interpret the results of a DGEA and to acquire understandings of the perturbations in a biological system. Biological pathways are sets of genes and their interactions forming a functional unit. DEGs help to identify pathways or networks that may be altered during a change of condition providing important information about diseases and its treatment process (Khatri et al., 2012). Pathway-based methods use predefined pathways or networks such as KEGG (Kanehisa & Goto, 2000) and Reactome (Fabregat et al., 2018), the expression measurements of the genes obtained from DGEA in combination with statistical methods and algorithms to identify specifically modulated pathways and processes (García-Campos et al., 2015).
Well established resources for pathway annotation are KEGG (Kyoto Encyclopedia of Genes and Genomes) (Kanehisa & Goto, 2000) and Reactome (Fabregat et al., 2018). KEGG pathways is a branch of KEGG database that hosts a collection of manually drawn pathway maps representing the molecular interaction, reaction and relation networks of cellular functions. Similarly, Reactome is an open-source, manually curated, peer-reviewed database for signaling and metabolic molecules with their interactions formed into different biological pathways for nineteen species (Fabregat et al., 2018). Both provide predefined pathways which are sets of genes and their interactions categorized into functional units. Starting from a gene interaction network, genes are labeled according to their role in a specific biological process. In this sense a particular gene can be assigned to different pathways. E.g., the human gene STAT1 is associated with 24 different pathways in the pathway annotation curated by KEGG and in 12 different pathways in the Reactome database. Although carefully produced, the assignment of genes to those predefined pathway units can be considered to be subjective to some degree and suffers from observational bias (Schnoes et al., 2013).
Existing pathway-based analysis approaches use different research designs, which can be categorized into ORA (Over-representation analysis), FCS (Functional class scoring) and pathway topology based methods. All of which aim to find a subset of genes, e.g., significantly differentially expressed genes, genes associated with a certain pathway more often than expected given the total set of examined genes, e.g. the whole genome background. ORA is the first and the most basic method of pathway analysis (García-Campos et al., 2015). It uses a DEG list with user defined cut-off for the log-fold change and pvalue (most commonly using absolute log-fold change ≥ 2 and p-value ≤ 0.05). Subsequently, sets of genes associated with annotated pathways are tested for being over-represented in the set of DEGs. To this end, hypergeometric distribution, chi-square tests, binomial probability or the Fisher’s exact test are used. Thereby the information of the topology of genes in the pathways is neglected (Bayerlová et al., 2015). Furthermore, ORA assumes that the biological pathways are independent of each other and ignores the fact that they cross-talk and overlap (García-Campos et al., 2015; Khatri et al., 2012).
Unlike ORA, FCS has no artificial cut-off to define a DEG list. FCS works in three steps. First it calculates the gene-level statistics including correlation of molecular measurements using differential expression of individual genes, ANOVA, t-test and Z-score. In the second step the statistics of individual genes in a pathway are transformed to an individual pathway-level statistic commonly using Kolmogorov-Smirnov statistic, mean or median. Finally the statistical significance of the pathwaylevel statistics is assessed. Although FCS covers some of the limitations from ORA, it still ignores the topology of genes in a pathway, cross-talk and overlap of the pathways (García-Campos et al., 2015; Khatri et al., 2012). Pathway topology based methods are similar to FCS except that they consider the topology of each gene during the gene-level statistics but still don’t aim to link different functional pathways (Khatri et al., 2012).
On these grounds we propose a novel approach to make use of the rich gene and protein interaction annotation resources available to gain additional functional insights from basic DGEA. To this, we start with the presupposition that expression of neighboring genes within a functional pathway are not independent from each other. Rather, they are often regulating each others expression or are part of the same regulon (Michalak, 2008). We understand that the categorization of links between genes into labeled pathways is often an arbitrary one, given the extensive cross talk between different pathways. Although these categories have proven to be useful in many situations, they force a certain perspective onto the interpretation of novel data. Based on these two principles, we aim to find sub-graphs of connected genes within cell signaling network which exhibit as a whole significant differential expression changes. This approach differs in two main aspects from common pathway analysis tools. First, it does not aim to identify functional pathways enriched in differentially expressed genes, but detects sub-graphs or branches in a network graph (potentially spanning more than one functionally grouped pathway) which is coherently modulated. Second, it aims to improve the DGEA on the gene level, by collecting the information of neighboring genes, which as a whole might exhibit prominent enough signal to be called, again as a whole, significantly modulated.
As input, information on functional links between genes provided by e.g. KEGG or Reactome and information on the differential expression status of single genes resulting from a DGEA, are required. As a result the analysis returns sub-graphs and their joint confidence scores, reflecting how the perturbation is migrated through the network. Furthermore, the entry points of perturbation in the networks and overlap with conventional pathway categories are returned. The output is prepared in a directed adjacency file, convenient for visualization, e.g., with StringApp (Morris et al., 2018), available as a Cytoscape plug-in (Shannon et al., 2003).
All of this can be helpful to understand the cause and effect of a stimulus and might inform about potential points of intervention. The proposed algorithm was implemented as a java program, which was named Modulated Sub-graph Finder (abbreviated MSF). MSF can help transform the information obtained from DGEA into comprehensible knowledge of signal transduction of genes and thereby being a valuable complement to existing pathway based methods. MSF is freely accessible from GitHub https://github.com/Modulated-Subgraph-Finder/MSF.
MSF is developed as a novel heuristic approach to find concertedly modulated sub-graphs in networks of biological interactions. MSF does not use predefined gene sets grouped into functional units, but rather relies purely on the network of interacting genes. The input network consists of nodes corresponding to genes and edges representing interactions. Furthermore it utilizes comprehensive results from a differential gene expression analysis to discover the sub-graphs, or modules, which are as a whole modulated.
MSF uses the individual gene’s p-values generated from the DGEA. The p-value expresses the probability that the null hypothesis of unmodified gene expression can’t be rejected for a given statistical model. To find significantly modulated sub-graphs individual p-values of the vicinal genes in the global network are combined into a single combined p-value, using a statistical method for combining dependent p-values described by Hartung (Hartung, 1998). Hartung’s method uses the inverse of standard normal distribution function, individual gene p-values are first transformed to their corresponding normal score. Then using these normal scores, the correlation between genes is calculated, the normal scores and correlation are applied to the inverse normal function to calculate the combined p-value for all genes examined, namely the examined sub-graph. The combined p-value of a sub-graph will express the significance of all genes in the sub-graph being modulated together. Thereby, the information from the different genes are used as, although not independent, replicated measurement of the behavior of the whole sub-graph. This potentially increases the power to detect also significant sub-graphs consisting of genes which are not significant on there own.
To reduce the complexity to score all possible connected sub-graphs MSF applies a four step heuristic as described in the following. The proceeding identification of modulated sub-graphs from a network by MSF is presented as a flowchart diagram (Figure 1).
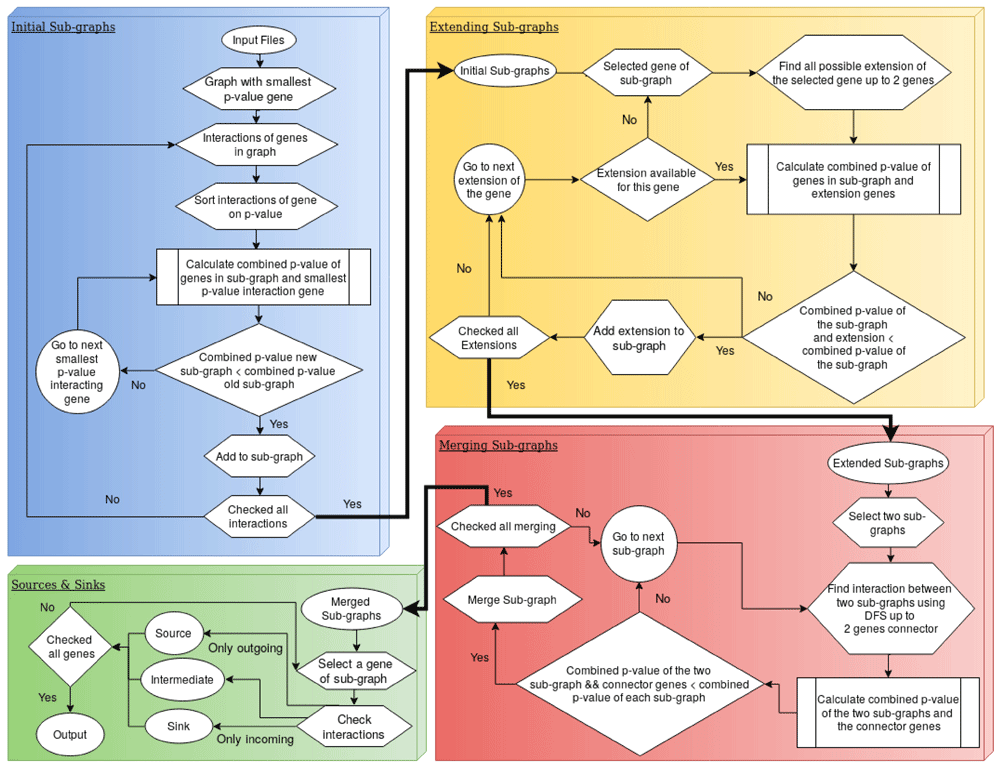
Initial modulated sub-graphs. MSF constructs the first sub-graph starting with the genes associated with the lowest (most significant) p-value deduced from the DGEA. From this seed it tries to extend the sub-graph by adding directly neighboring genes, starting with the most significant one. A single combined p-value is calculated for the extended sub-graph. If the combined p-value is smaller than the p-value of the original sub-graph, the extended sub-graph is accepted. This step is iteratively repeated until no further extension is accepted. In this case the process starts over with all remaining genes not yet in a significantly modulated sub-graph. This step identifies all simple sub-graphs that are modulated in the whole network.
Extending modulated sub-graphs. In the next step, we check if any of the initial modulated sub-graphs could further be extended by adding more than one gene at a time. This is done by testing all possible extension paths up to N (default 2) genes at all nodes in the sub-graph. Again, this step is iteratively repeated until no further genes are added to the significant differentially expressed sub-graphs. This step bridges small gaps of genes without a clear differential signal in the DGEA.
Merging modulated sub-graphs. After detection and extension of the modulated subgraphs, they are tested if combined sub-graphs score better than on their own. The merging of the sub-graphs is done by depth first search traversal from the first sub-graph to the second sub-graph. If the two sub-graphs merge with the connector of at most N genes (default 1 gene) and the combined p-value of the merged sub-graph including the bridging genes in between is less than the individual p-values of the two sub-graphs, the two sub-graphs are merged together to one big modulated sub-graph. This step is repeated iteratively until no sub-graphs could be merged.
Finding sources & sinks. In a last post processing step MSF identifies the trigger points of the modulated sub-graphs. These trigger genes are the sources of the sub-graphs with only outgoing edges. These genes can be interpreted as the possible entry points of perturbation from where the stimulus causes downstream effects. In the same spirit the most downstream genes of the modulated sub-graph are identified and defined as sinks. Sinks can be interpreted as the effectors where the integrated information within the signal transduction network is set to action. Due to circular loops not all sub-graphs are guaranteed to have sources or sinks.
Operation. MSF requires Java version 8 and JDK 1.8. The few package dependencies are already been added to the release. MSF runs fast on a standard laptop computer and so it has normal system requirements. To run MSF, the user must provide two text files, one containing the DGEA and the second one containing the interactions in an adjacency format file. Example files and a detailed tutorial to use MSF has been provided on github https://github. com/Modulated-Subgraph-Finder/MSF.
To demonstrate its usefulness, MSF is applied to an RNA-seq data set of primary human monocytederived macrophages (MDMs) infected with Ebola virus (GSE84188) (Olejnik et al., 2017). Ebola Virus (EBOV) belongs to the Filoviridea family; filamentous, enveloped and single stranded RNA viruses. EBOV causes hemorrhagic fever in humans, inducing the host innate and adaptive immune response to be unable to control virus infection (Prins et al., 2009). Until now, there are no approved antiviral drugs for the treatment of Ebola virus infection (Konde et al., 2017; Rhein et al., 2015). The initial targets of EBOV are the macrophages and dendritic immune cells (Falasca et al., 2015; Rhein et al., 2015). EBOV inhibits the critical innate immune response of the host, which includes the activation of alpha/beta interferon (IFN-α/β) (Cárdenas et al., 2006; Konde et al., 2017; Prins et al., 2009). It has been proposed that IFNα/β should be tested against Ebola for its antiviral activity through clinical trials (Konde et al., 2017). Ebola infection data was selected to test the approach because it has been well recognized for the last several decades, and vast literature is available for the pathogenesis of Ebola. Thereby facilitating the verification of the results of MSF with the vast literature present on Ebola infection. Especially, the detection of IFN-α/β as point of action for the virus, could be considered as a basic indicator of the correctness and usefulness of the approach.
EBOV infection count data was downloaded from Gene Expression Omnibus (GEO) (GSE84188). Differential gene expression analysis was performed on the count data with edgeR package (version 3.4.2) (Robinson et al., 2010). The DEG analysis results generated by edgeR were used as input for MSF. Directed cell signaling interactions were filtered from Reactome Functional interactions (FIs) Version 2016 (Wu et al., 2016), which was used as a second input for MSF.
The EBOV infection experiments describe the course of infection at three time-points 6, 24 and 48 hour post infection (hpi). For the earliest time point at 6 hpi, three large modulated sub-graphs were identified with 41, 107, and 18 genes were identified, as well as two with less than 4 genes. The modulated sub-graphs consist predominantly of cytokines, chemokines (CXCL10, CCL8) and Interleukin genes (IL6, IL27, IL23). IFNB1 and IFNA1 were both identified as two of the possible sources in one of the sub-graph identified with 18 genes. Most of the sources identified by MSF were type I interferon induced genes (Supplementary material 6H). At 24 hpi eight modulated sub-graphs were identified with four main sub-graphs consisting of 27, 101, 134 and 194 genes, others being smaller than 10 genes. IFNB1 and IFNA1 were identified as the two sources out of the total sources. For the last time-point 48 hpi, six modulated sub-graphs were identified. Three of the sub-graphs were less than nine genes and main sub-graphs had 122, 191 and 202 genes. IFNB1 was identified as one of the sources in the most significantly modulated sub-graph (Supplementary).
As stated earlier IFN-α/β was reported to be one of the target genes of Ebola infection. We were able to successfully identify IFNA1 as a source in all three time-points and INFB1 in two of the time-points. Although IFNA1 and IFNB1 were already among the most significant gene in the DGEA during the later time points, MSF was also able to detect them as a source in the very early time-point when the genes were not significant based on the individual DGEA alone. Identifying the possible sources will reduce the search space for potential target genes and can help the biologist as the starting point of clinical testing for drugs and vaccines against an infection.
Table 1 compares the results of MSF, namely the number of detected sub-modules and their total genes numbers, to a simple analysis of mapping significantly modulated genes from the DGEA to the network and joining neighbors to modules. The numbers indicate that MSF detects less but larger sub-modules, applying its statistical test. Furthermore, the dependency of the results from the p-value cutoff choice is demonstrated for the DGEA, which is avoided for MSF altogether.
hpi - hours post infection.
Three main modulated sub-graphs identified by MSF at 6 hpi are shown in Figure 2. The gene based graphs on the right hand side, represent the immediate output of the MSF-analysis, visualized by StringApp (Morris et al., 2018) in Cytoscape (Shannon et al., 2003). Each node represents a gene part of a modulated sub-graph, whereby the associated colors code the functional annotation deduced from KEGG Pathways. The cross-talk between the pathways and also the multiple employment of many genes is evident. The more schematic drawing on the right side represents the effortlessly deduced flow of information between the sensors and effectors in this particular example.
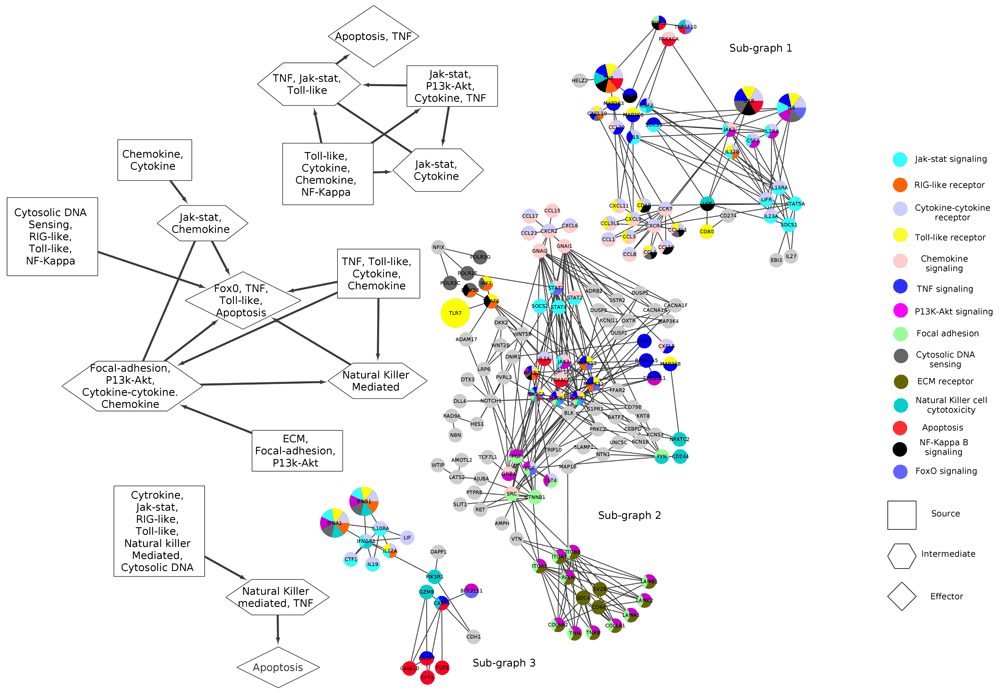
The node coloring is associated to KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways referring to the colors in the legend. The graph edges are from Reactome. Important genes to EBOV infection as from literature are enlarged in the graph.
In more detail, sub-graph 1 (top) shows how the activation of toll-like receptor, cytokine, chemokine and jakstat genes lead via TNF into apoptosis. The next significant sub-graph (sub-graph 2: middle) reveals how information from the Extra-cellular matrix (ECM) receptor, which are reported to interact with Ebola glycoprotein (GP) (Veljkovic et al., 2015), chemokines and cytokines, and cytosolic DNA sensing, is integrated into again modulation of apoptosis pathway. Eventually, sub-graph 3 (bottom) demonstrate how INFA1 and INFB1 modulate once more, via only a few intermediate steps, the apoptotic response of the cell.
This show cast example demonstrates with how little effort complex data can be interpreted, help to apprehend the dynamics of the underlying processes and suggest testable hypothesis and potential points of intervention.
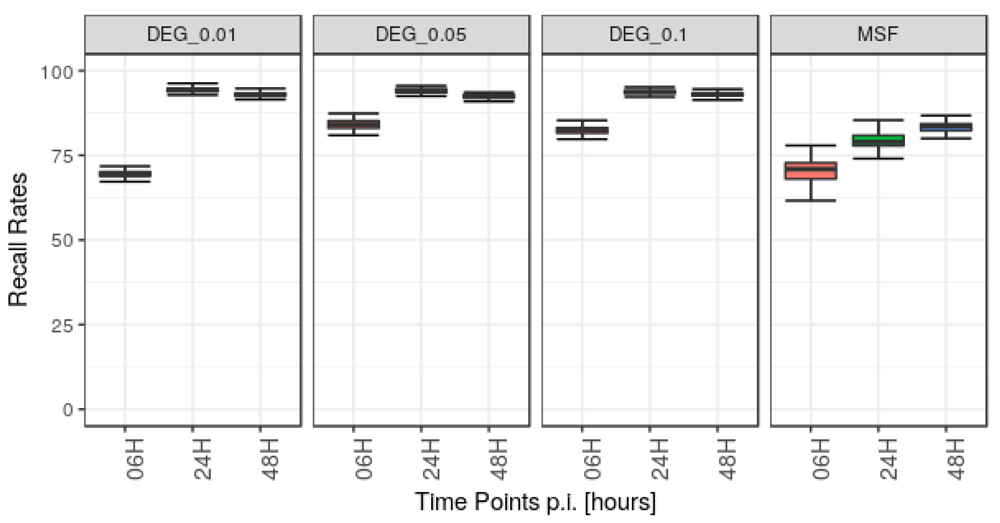
A potential concern is how noise in the gene expression measurements affects our analysis. To assess the robustness and stability of our method, we therefore added Poisson distributed noise to the read counts of the three time-points data set, used above. Then DGEA was carried out on the disturbed data with the same parameters as for the native data using edgeR, followed by analysis with MSF. This procedure was carried out 100 times. Every time the genes from the modulated sub-graphs identified from noisy data were compared to the genes of sub-graphs identified from the native data. This was also done for the DEG obtained for each run, using three different cutoffs of FDR 0.01, 0.05 and 0.1. The robustness of MSF and the DEG analysis for the time-point 6, 24, and 48 hpi are shown in Figure 3. The procedure for how data noise was modeled can be considered as rather stringent, which is already reflected by the limited recall rate in the edgeR based DGEA, between 68 % (6 hpi) and 93 % (24 hpi). For MSF-analysis the observed median recall rates lay between 71 % (6 hpi) and 84 % (48 hpi). The better performance of the pure DGEA can be explained by the fact that disturbed p-values do not change the results for DGEA as long as the p-value does not rise above the chosen cutoff value. In contrast, MSF is sensitive to p-value changes across the whole range of possible values.
Gene enrichment analysis was performed using Reactome Analyze data tool (Fabregat et al., 2018). Reactome’s over-representation analysis tests whether certain Reactome pathways are enriched by the list of genes submitted. Genes from MSF identified sub-graphs for each time-point were analyzed for gene enrichment analysis. For comparison the DEG results from edgeR were filtered using three different cut offs of adjusted p-value 0.01, 0.05 and 0.1. This three subsets of DEG list were used for gene enrichment analysis. The compression of MSF and the three subset DEG list is shown in Figure 4.
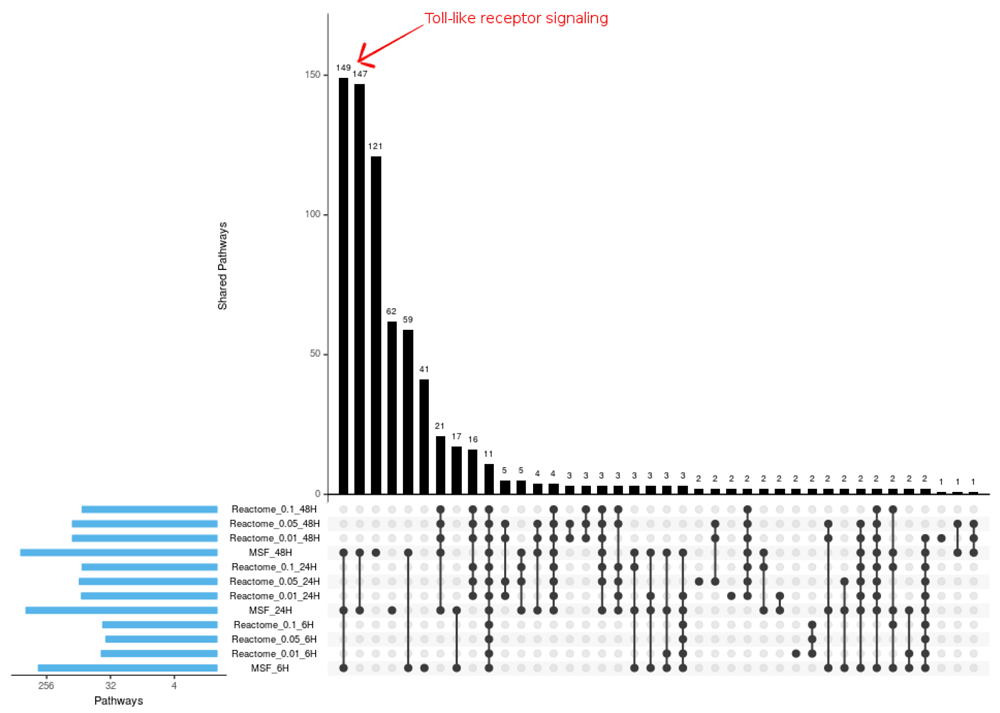
All the different toll-like receptor signaling pathways identified from Modulated Sub-graph Finder (MSF) identified genes are marked.
The comparison shows most of the pathways known from literature to be dis-regulated by Ebola infection are enriched in both the enrichment analysis. Toll-Like receptor signaling pathway when interacts with EBOV glycoprotein (GP), it triggers the activation of cytokines (Olejnik et al., 2017). Toll-like receptor pathway is expected to be dis-regulated in the early stage of infection, this pathway was not identified as significantly dis-regulated when p-value cut off DEG lists were analyzed for enrichment. Ten toll-like receptor cascades were identified as dis-regulated from gene enrichment analysis of MSF identified genes, not a single one of these pathways was shown to be dis-regulated in DEG cut off lists. Since MSF considers the complete DEG results, even the weak signal at the earliest time-point was detected; for example Toll-like receptor signaling.
Classic pathway analysis tools aim to detect in lists of significantly deregulated genes enriched associations with pathway genes categorized by their biological function and their interactions. Thereby, depending on the tool, the internal pathway topology is considered or neglected all together. The here presented tool, MSF, employs a different approach, by aiming to detect sub-graphs in whole gene regulatory networks which are significantly deregulated in a concerted manner. To this end, neighboring genes in the user provided network are tested for jointly common regulation. Exploiting that each gene’s abundance, although not independent from its neighbors, is measured repeatedly on its own, sensitivity can be increased by our applied p-value meta-analysis, namely Hartung’s method. This potentially enables to call nonsignificant modulated genes based on the DGEA, to be convincingly part of a deregulated gene group. Furthermore, it allows to identify connected sub-graphs, representing the propagation of gene regulation perturbation in the input network. A better understanding of this propagation, especially the critical spots such as sensors, effectors, or intermediate bottlenecks and hubs, facilitates the projection of potential intervention points, e.g., for drug development. Since MSF only uses interaction information in gene regulation networks, but not the functional grouping of the genes into functional pathways, it is especially adapted to discover so called cross-talk between such pathways.
MSF is a fast, robust and easy to use tool to find concertedly modulated sub-graphs in a given network. Its implementation in JAVA enables its use across many operating systems. It requires as input the results from a differential gene expression analysis in the appropriate file format. So far the raw output from edgeR (Robinson et al., 2010) and DESeq2 (Love et al., 2014) are supported. Furthermore, a gene network has to be provided in the format of a directed adjacency list.
The Ebola infection RNA-seq data set analyzed during the current study are available in the GEO repository (GSE84188) (Olejnik et al., 2017). The cell signaling network file used is from Reactome Functional interactions (FIs) Version 2016 (Wu et al., 2016).
Source code is available from GitHub: https://github.com/Modulated-Subgraph-Finder/MSF
Archived source code at time of publication: http://doi.org/10.5281/zenodo.1400242 (Farman, 2018).
Software license: MIT license.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)