Keywords
Chlorophyta, Lake Baikal, Chloroplas Genome, Genetic Polymorphism, Mutation Rate
Chlorophyta, Lake Baikal, Chloroplas Genome, Genetic Polymorphism, Mutation Rate
Lake Baikal, located in Southeastern Siberia, is the largest by volume and the oldest great lake on the planet, and several signs of ecological crisis in Lake Baikal have been observed since 2010–2011 [Bormotov, 2012; Khanaev et al., 2018; Kravtsova et al., 2014; Timoshkin et al., 2016]. One of these signs is severe disease and death of sponges which is now observed in almost all parts of the lake. The symptoms of the disease begin with the appearance of pink and brown spots on the surface of the sponge and terminate with complete destruction of the sponge tissues. The cause of the disease is still unclear.
Endemic freshwater Baikal sponges (Demospongiae, Lubomirskiidae) dominate their biomass among the benthic organisms of the littoral at depths from 3 to 25 m covering 47% of the available surfaces [Pile et al., 1997]. In healthy condition sponges have a green color, mainly explained by the presence of a photosynthetic symbiont, an intracellular coccoid green algae. This algae belongs to the Cholorophyta division, and is close in taxonomy to the Choricystis genus [Chernogor et al., 2013]. It is natural to assume, that the photosynthetic symbiont is the source of feeding for sponge cells. And, the change of color of sponge tissue could indicate that the chloroplasts of the symbiont are damaged in the early stages of the disease. So, precise study of this algae symbiont could be of critical importance in investigating the cause and consequences of sponge disease.
The sequencing and comparative analysis of chloroplast DNA is a conventional method for detailed study of planctonic algae [Lemieux et al., 2014; Lemieux et al., 2015]. Normally, chloroplast DNA sequences are determined using cultivated algae and de novo assembly of genomic DNA reads [Twyford & Ness, 2017]. But for uncultured species, the chloroplast genome can be obtained using metagenome sequencing [Worden et al., 2012]. For symbiotic algae from Baikal sponges, this strategy is probably more efficient, and the comparative analysis of samples of healthy and diseased sponges can provide a deeper look into the features of chloroplast genome affected by sponge disease. The presence and properties of polymorphic sites on the chloroplast genome could be an effective way to investigate the variations of genome sequence depending on the disease state of the sponge. The study of the distribution of bacterial strains depending on geographic location [Truong et al., 2017] can be mentioned as a precedent, where gene-batteries typical for gut microbiome were compared using the distribution of polymorphic sites in genome sequences, using metagenome sequencing.
Three samples of freshwater sponge Lubomirskia baicalensis were collected from Lake Baikal in the Bol'shiye Koty area (51° 90´ 69 N ´´, 105° 07´ 05 E´´) at a depth of 10 m by scuba divers in June 2016. One sample was obtained from the sponge that was healthy in appearance (exhibiting a green colour), one sample was taken from diseased sponge and one from dead rotten sponge tissues. The collected samples were immediately placed in containers with Baikal water and ice and transported to the lab, maintaining a constant water temperature. For all three samples Illumina pair-end reads were obtained by DNA metagenome sequencing in Novogene Inc. (Illumina PE 150). The extraction and sequencing of RNA in the samples was also performed at Novogene Inc., to represent their metatranscriptome content. This was possible only for healthy and diseased sponge tissues; not enough RNA was extracted from the rotten tissues. The DNA metagenomic reads were processed by a conventional bioinformatics pipeline implemented at Novogene Inc, including the filtering of sequencing errors and the assembly of contigs using SoapDeNovo assembler [Luo et al., 2012]. RNA metatranscriptomic reads were filtered and trimmed using Trimmomatic 0.35 software [Bolger et al., 2014].
The sequence of chloroplast genome Choricystis parasitica (NC_025539) was used as a template for assembly and for comparative analysis because it is the chloroplast genome closest to the available genomes. For the assembly of the targeted genome, the following steps were performed:
1. Scaffolds were obtained by de novo assembly of each DNA metagenomic sample by conventional utilities with the use of the SoapDeNovo assembler
2. Scaffolds were aligned to the reference sequence of chloroplast using Blastn (evalue threshold 1e-40)
3. The paired-end sequence reads from 3 DNA metagenomics libraries and 2 RNA metatransriptomic libraries were aligned to the selected scaffolds using Bowtie2 (v. 2.2.6)
4. The aligned reads from all samples were collected and assembled de novo using the Inchworm assembler from the Trinityrnaseq 2.6.5 package [Grabherr et al., 2011] with the lowest possible tolerance to sequencing errors and the highest possible value of k-mer size (K=31)
5. The contigs obtained after the de novo assembly by Inchworm were compared with the reference genome; this allowed the selection of a single contig of length 55638 with high homology to the reference genome. It was the only contig which could be reliably identified as a fragment of the chloroplast genome.
Open reading frames were identified in the obtained contig, and most of the identified proteins are annotated following the annotations of the reference genome. TrnaSCAN 1.4 software [Lowe & Eddy, 1997] was used for identification of the transport RNA in the putative chloroplast sequence, and the locations of 18S rRNA and 23S rRNA were identified by a direct alignment with reference rRNAs using the Mummer 3.23 package [Kurtz et al., 2004].
In order to separate it from traces of sequencing errors, the selection of the polymorphic sites in the genome was implemented following the approach described in [Truong et al., 2017]. Each of the RNA and DNA samples represented as pair-end reads was separately aligned to the assembled fragment of the chloroplast genome using Bowtie2; the alignments were then processed using the Samtools 1.7 software pipeline with conventional settings, and the approach proposed in [Truong, 2017] was used to identify the polymorphic sites.
Describing the algorithm, for each position s on the alignment of the reads against the Ns is defined as the total number of reads covering it, and Ts is defined as the number of reads supporting the most abundant allele. Given the sequencing error rate E, the non-polymorphic null hypothesis was rejected if the probability that the number Ns − Ts of reads coming from the non-dominant allele is <α = 0.05. This is estimated using the probability mass function of a binomial distribution with Ns trials and the successful rate 1 − E. The error rate was set to 0.01 for Illumina sequencing. The bases with quality below 30 were removed and the reads with an average identity to the reference below 99% were ignored before applying the statistical test. Failing to reject the null hypothesis reflects the absence of alternative alleles or inability of distinguishing between low-coverage potential alternative alleles and sequencing noise.
Thus, the number of polymorphic sites could be counted for each gene. Another property of each gene is the number of polymorphic sites where the count of alternative alleles is higher than the count of dominant allele (Ts < Ns - Ts). This property could detect mutations in the sample genotype and phenotype, for each gene.
The chloroplast genome sequences of Picocystis salinarum (NC_024828), Myrmecia israelensis (KM462861), Botryococcus braunii (KM462884), Coccomyxa subellipsoidea (NC_015084), Hydrodictyon reticulatum (NC_034655), Mychonastes jurisii (NC_028579) and Chlorella vulgaris (NC_001865) were used for a reconstruction of the phylogenetic trees for the 16S ribosomal RNA (rrs gene) and the ATP synthase subunit beta (atpB gene). The nucleotide sequences of the selected genes were aligned using Mafft 7.27 software [Katoh & Standley, 2013]. The trees were constructed using the FastMe 2.1.5.1 software [Lefort et al., 2015], with the distance-based neighbor-joining method to select tree topology and Jukes-Cantor measure to calculate the distances between genes.
Analysis was performed using custom scripts in Python 2.7 (see Data and software availability section).
The chloroplast genome of the Choricystis parasitica algae is a circular DNA 94206 base pairs in length. The comparison of open reading frames of the candidate genome fragment from the metagenomic samples with annotated genes of C. parasitica support the statement that this genome fragment of length 55638 is a large part of the chloroplast genome of algae close to the C. parasitica species. Figure 1 illustrates the order of genes in these two related chloroplasts. The comparison of gene sequences shows them to be up to 98% identical in these two species.
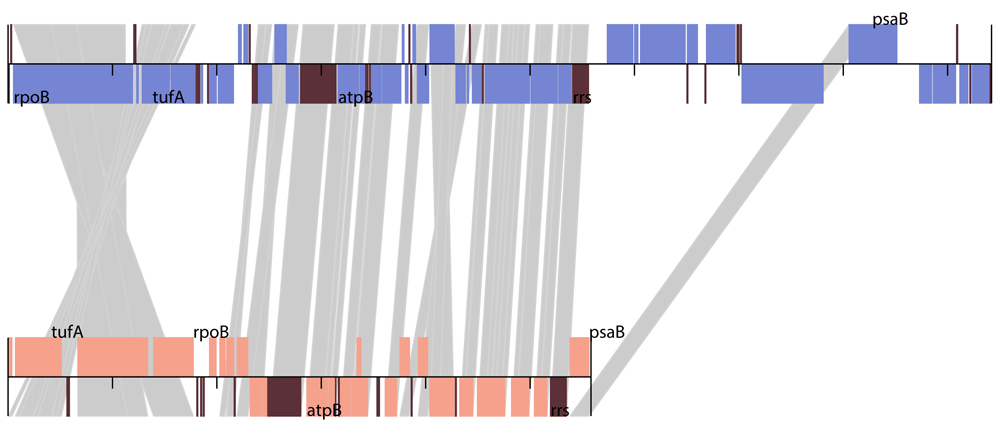
Upper track: C.parasitica chloroplast; Lower track: Chloroplast of sponge symbiont. The start position of the C.parasitica chloroplast sequence was changed to fit the location of the fragment shown at the bottom. The rRNA and tRNA locations are shown in brown color for both tracks. Text labels show the locations of several selected genes in both genomes.
The phylogenetic trees for the two selected genes, 16S ribosomal RNA and ATP synthase beta (Figure 2) in general confirm the conventional relations between Cholorophyta algae [Lemieux et al., 2014; Lemieux et al., 2015]. Figure 2 suggests that the symbiotic algae of L. baikalensis sponge is close in taxonomy to the Choricistys genus.
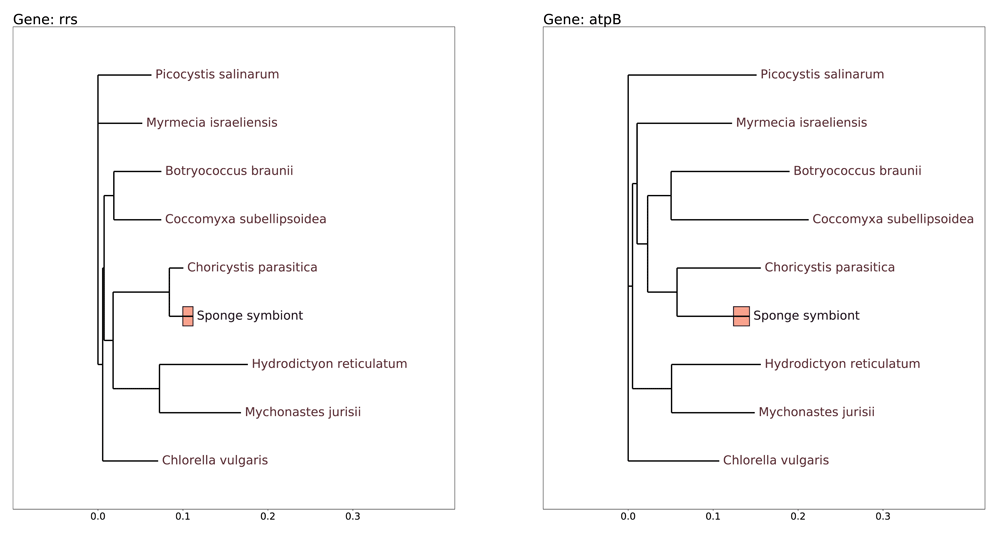
Bars located at the node for the studied chloroplast genome represent the relative number of polymorphic positions, in all 5 studied samples, at the 1:1 scale.
The bars on Figure 2 which show the proportion of polymorphic positions in the genes of symbiotic algae in metagenomic samples is comparable in scale with the distances between genera. This observation needs discussion, because a timescale which separates the origins of the close genera in Figure 2 implies a much larger timescale than that which could characterize the separation of the chloroplast strains detected in the metagenome. Partially this can be explained by the RNA editing and similar modifications which lead to the accumulation of polymorphic positions.
A different view of the unexpectedly high proportion of polymorphic sites in the metagenomic samples is illustrated in Figure 3. Here, the proportion of polymorphic sites, and the proportion of polymorphic sites with a low abundance of dominant allele (“mutations”) is shown separately for each DNA and RNA sample. The results of Figure 3 are presented separately for each gene frame, and for a whole set of genes.
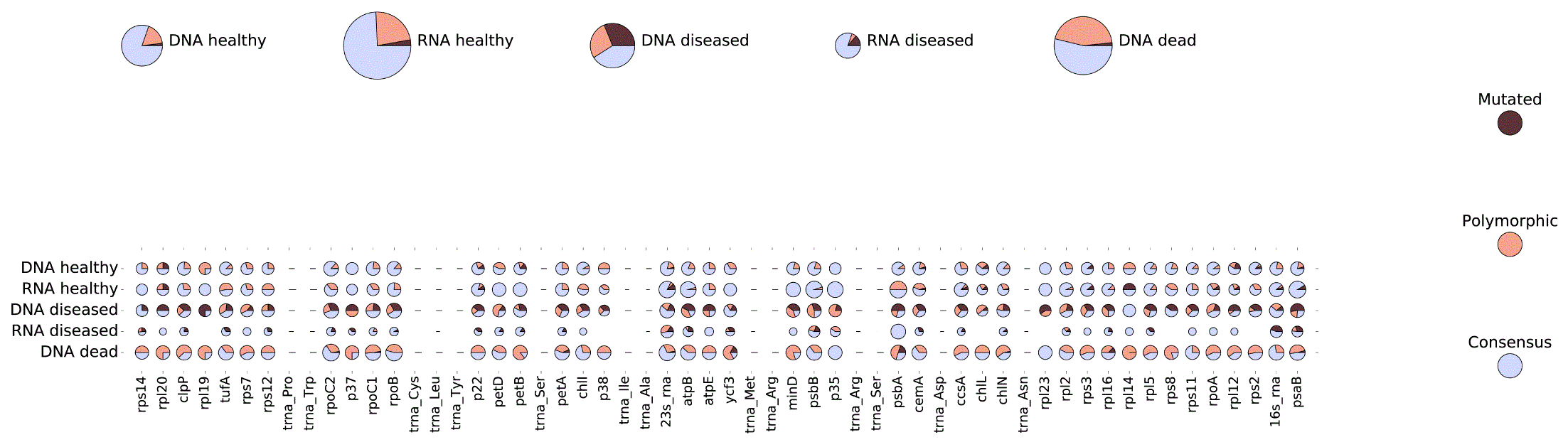
The results for each of the annotated genes are show in the bottom part. The upper line presents the integrated results for each sample. The proportions of polymorphic sites and the sites with high levels of alternative alleles (“mutations”) are shown as pie charts, relatively to a total number of polymorphic sites in all samples. The proportion of sites which are polymorphic in some other samples, but not in the given sample, are shown in light blue. The legend on the right shows a color scheme used to represent three types of sites. The circle radius represents a total number of sequencing reads aligned to the gene segment and used to identify polymorphic sites. The scale of the circle radii is transformed for better appearance, to compensate for the high variations in the numbers of aligned reads.
The proportion of polymorphic sites in the DNA and RNA metagenomes for the sample of healthy sponge tissues reflects the natural situation, where the quantity of matrix RNA in the chloroplast organelle is in general higher than the quantity of DNA. Here the polymorphic sites in the DNA sequences may arise due to natural heterogeneity of chloroplast genomes, but the dominant strain is clearly identified. The number of polymorphic sites in the RNA sequences is slightly higher than in DNA sequences due to RNA editing and other modifications.
In contrast, in the DNA and RNA samples of the diseased tissue, the quantity of chloroplast DNA is decreased, and the quantity of RNA is decreased even more, reflecting the fact of disease and the low level of chloroplast activity. Importantly, the number of polymorphic sites is sharply higher in the remaining DNA sequences, and the alternative alleles are presented in high proportion. And, for RNA sequences, the dominant allele is present much less than alternative alleles. For the case of dead tissue, where RNA couldn't be extracted, the discussion about the observed number of DNA molecules and the proportion of polymorphic sites is beyond the scope of this study.
The natural assumption about the diseased but alive tissue is that living cells are desperately trying to survive. Adaptation to a changed environment is the one of the best ways to survive. The accumulation of mutations is a straightforward form of adaptation, and this could be confirmed by the results of Figure 3 for the sample of diseased sponge tissue. The rapid increase of mutations in the genome can be observed in the chart for chloroplast DNA, and as it may be suggested from the chart for chloroplast RNA, that the mutations which help survival are fixed in the cells which still continue to develop.
The observed signs of extensive mutations in response to severe stress are somewhat controversial when compared to a the widely accepted concept of molecular clocks and a theory of neutral evolution [Kimura, 1968; Margoliash, 1963; Zuckerkandl & Pauling, 1962] where mutations are appear randomly and are independent from the environment. But in several studies the presence of adaptive mutations in response to stress has been detected in certain species, as reviewed in [Rosenberg, 2001; Wright, 2004]. So the present result cannot be treated as completely inadequate, in conditions of severe and unusual crisis of the whole ecosystem.
The signs of the large-scale ecological crisis in Lake Baikal are confirmed from many sources, and ecological crises on such a large scale are rare in the documented history of water ecosystems. Sequencing technologies have appeared only in a recent years, and, to the authors best knowledge, no cases of large and sharp changes in ecosystems have been documented using the tools of molecular biology.
The importance of Lake Baikal itself as an ecosystem with an unusual diversity of endemic species, and as a glorious source of pure drinking water, is a subject high above the economic and pragmatic reasons which are usually considered in molecular biology studies. However the present results suggest that the conventional approaches of molecular biology may be insufficient to adequately describe situations of ecological crisis. In particular, the observations of rapid accumulation of mutations in the chloroplast genomes in the diseased tissues could indicate that the concept of molecular clocks is inappropriate in rapidly changing ecosystems.
What’s more, using the tools of molecular biology to study the Baikal ecosystem has another importance; it is a unique chance to accumulate observations about a rapidly changing environment. Great lakes are themselves simplified cases of large-scale marine ecosystems. The presented results, as a part of all Baikal ecosystem studies, could find application, not only in the challenge of minimizing the consequences of the crisis in Baikal, but also in the possible future global challenges caused by sudden changes in ecosystems of any scale.
In particular, the reconstructed genome of the symbiotic algae may improve knowledge about a cause of sponge disease, and indirectly narrow the possible strategies to prevent the spread of destruction in the Baikal ecosystem. The presented description of the genome may be helpful in the evolutionary studies of marine and freshwater Cholorophyta algae.
The nucleotide sequence of the chloroplast genome fragment is deposited to Genbank under the accession number: MH591948
Nucleotide sequences have also been deposited with the European Nucleotide Archive (ENA) of the European Molecular Biology Laboratory-European Bioinformatics Institute (EMBL-EBI) under study number: ERP110335.
The sequencing reads and source codes of scripts sufficient to reproduce the presented results are available from GitHub: https://github.com/sferanchuk/bsponge_chloroplast
Archived source code at time of publication available at: https://doi.org/10.5281/zenodo.1326765 [Feranchuk, 2018].
(License: CC BY 4.0).
Custom scripts on Python (v 2.7) were used to run the pipeline and present the results. Python libraries pysam (0.14.1), biopython (1.66) and matplotlib (2.2.2) are required to run the scripts.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)