Keywords
High throughput sequencing, whole exome sequencing, RNA sequencing, variant analysis, single nucleotide variant, R, Bioconductor
This article is included in the RPackage gateway.
This article is included in the Bioconductor gateway.
High throughput sequencing, whole exome sequencing, RNA sequencing, variant analysis, single nucleotide variant, R, Bioconductor
Based on reviewers comments current version of the manuscript, the software and its documentation have been further improved. The manuscript clarifies the purpose and functionality of seqCAT as a software for genotype analysis. The software has been revised to allow access of information from additional file formats. The structure and use of data objects have been streamlined to only utilise data frames at the user-level. Software improvements aim to simplify the use of seqCAT and clarify the documentation. All changes implemented are described in the manuscript.
See the authors' detailed response to the review by Jean Fan
See the authors' detailed response to the review by Matej Lexa
High throughput sequencing (HTS) technologies such as genome, exome and RNA sequencing (RNA-seq) have become some of the most powerful and widely used tools in biological research worldwide, and an increasing amount of such data is being stored in online data repositories (e.g. the Gene Expression Omnibus, GEO, and the Sequence Read Archive, SRA)1–3. While decreasing experimental costs and optimised protocols enable a broad range of researchers to apply HTS to their respective scientific questions, e.g. gene expression or genetic variation, the analysis of the resulting data is not a trivial matter, often requiring a high level of bioinformatic expertise4,5. This is especially true for variant analyses, where data is commonly stored using the relatively complex variant call format (VCF). There is a multitude of software packages that can analyse data in the VCF format, but they vary in their functionality, outputs and simplicity of use. Software that focuses on applications other than genetic heterogeneity or general analysis of HTS variant data include Anvi'o6 (metagenomics of microbial populations), PhyloSNP7 (phylogenetic trees from SNP data), KING8 (kinship estimation), SomaticSniper9 (comparisons of paired tumour and normal samples), PLINK10 (a toolkit for analysing whole genome association and population data), and vcfR11 (quality control and filtering of VCF files). Many of these require use of the command line and are no longer being actively developed. Two examples of command line-based software are vcftools12 and the R-package VariantAnnotation13. Both of these provide underlying structure and several ways to analyse variant data, but require further analysis of their outputs and can be difficult for less experienced users to work with. Software such as BEDTools14, BEDOPS15 and related tools that work on genomic interval-based comparisons similarly require additional downstream analyses and necessitate using a command line-interface. While some of the previously mentioned software allow for a considerable number of different analyses to be performed, the choice of analysis or how to perform it based on the biological question at hand is not always apparent. The necessity of downstream analyses is also an important consideration. There are also several software tools with a more easy-to-use graphical interface such as the Integrative Genomics Viewer16 or the web-based Ensembl Genome Browser),17. While such software can more easily provide visualisations and figures, they are generally more limited in their functionality. Web-based applications are restricted in the amount of data that can be uploaded, and also come with the added issue of data security18. Proprietary software (such as the Ingenuity Variant Analysis)19 not only require a licence to use, but also constitute a “black box” where the underlying methods are not available for direct inspection or scrutiny.
There is thus a need for transparent, user-friendly and powerful bioinformatic tools to enable as many researchers as possible to analyse, visualise and interpret their own and publicly available HTS data. Two important aspects of such analyses is the true identity of cells analysed and comparability of both the biological samples and the data sets. Validation and evaluation of cell line authenticity, for example, is an increasingly widespread issue, as is the question of biological equivalence for any sample in general20. Here we present an open source R-package, the High Throughput Sequencing Cell Authentication Toolkit (seqCAT), which uses data from HTS experiments (whether it be of DNA or RNA origin) to investigate these matters.
One of the common outputs from HTS experiments is that of sequence variation. Single nucleotide variants (SNVs), for example, are sequence variations at the nucleotide level. Such data is the output of many variant calling programs and algorithms, which is used by seqCAT in order to analyse genetic differences between samples. We have previously demonstrated the usefulness and general applicability of such analyses for both cell line authentication21 and genetic heterogeneity in public cell line datasets22. The capabilities of seqCAT include creation of SNV profiles from VCF files, comparisons of the overall genetic similarity between profiles, investigations of SNV impact distributions (i.e. variants’ predicted impact on protein function) as well as interrogations of the genotypes of previously known or user-specified variants across samples. Each individual profile can represent SNVs from a HTS experiment or from an external variant database.
The seqCAT package distinguishes itself from those previously mentioned several ways. First, it includes not only processing and filtering of variant data, but also a number of downstream analyses and visualisations. It provides a simple way for researchers to explore, subset and group variants in ways that are of biological importance. Furthermore, its implementation in the R programming language allows the user to work on their data without the command line, while also giving access to the extensive library of packages provided by the R environment. While its applications are not as numerous as some existing software, seqCAT is focused in its exploration of genomic and transcriptomic variation across many biological contexts. Finally, seqCAT contains a detailed manual, only presents the user with base-R data objects and contains several helper functions aimed solely at making it simple and easy to use, allowing even novices to utilise it.
In the present study, we use seqCAT to explore genetic differences within a public dataset containing both whole exome sequencing (WES) and RNA-seq data for long-term organoid cultures. We show that the organoids are genetically stable over a culture-period of several months, corroborating the original authors’ conclusions. We also demonstrate how seqCAT can be used to compare DNA- and RNA-based variant calls using the same dataset. The results highlight potential uses of variant analyses and demonstrate how seqCAT may be utilised to interrogate genetic differences at both the global and gene-specific level.
SeqCAT was developed for the Bioconductor repository for R-packages. It follows existing best coding practises, including a clean, modular and robust design, as per the requirements for Bioconductor packages23. The basis of all seqCAT analyses are SNV profiles: collections of filtered, high-quality SNVs for any given sample. The creation of these SNV profiles is performed by filtering an input VCF file based on the available variant calling quality metrics21. These criteria are taken directly from the input VCF; they are based on the variant calling software used to create them and are not specific to seqCAT. There is also an option to skip this filtering step, for cases where the VCF does not contain any filtering information from the variant caller or when the user does not wish to perform filtering. Additional optional filtering steps include removal of variants below a specified sequencing depth (ten by default), removal of mitochondrial and non-standard chromosomes, as well as removal of duplicate variant entries. While profiles for individual samples may be created as needed by the user, several convenience-functions for working with multiple VCFs and profiles in aggregate are also available. SeqCAT can analyse VCF files with or without annotations from e.g. snpEff24.
The SNV profiles are subsequently compared to each other in a pairwise manner, yielding information on e.g. the overlap (SNVs that are present in both samples being compared), the concordance (the proportion of SNVs with identical genotypes for both samples) and the similarity score (a previously defined weighted measure of the concordance)22. Comparisons may be performed individually or in aggregate, depending on what type of analysis the user is interested in. Comparisons with external databases is also possible; seqCAT currently contains functionality to read and compare variants present in the Catalogue of somatic mutations in cancer (COSMIC) database25. Only overlapping variants are analysed by default, but non-overlaps can optionally be included as well. Examining specific chromosomes, genes or genomic regions is also possible, as are analyses of variant functionality through their predicted impact on protein-function.
Installation of both seqCAT and its dependencies is simple, and its use is described in-depth in its vignette; a major design goal of seqCAT was ease-of-use for a broad range of researchers, regardless of expertise in R. While existing data structures and objects from Bioconductor are used internally, none of these are required learning for the user; results are given as standard R-objects13,26. This makes exploration of the data as simple and easy as possible for the user. SeqCAT allows for re-analysis of already created SNV profiles, facilitating comparisons of samples across any number of datasets and includes several functions for creating publication-ready figures. All these capabilities make seqCAT a useful, simple and intuitive tool for a wide range of researchers.
To demonstrate the capabilities of seqCAT, we analysed a recently published dataset from Broutier et al.27. The authors created liver cancer-derived organoids for modelling disease and performed both whole exome sequencing and RNA-seq on the original tissues and the organoid cultures. We used seqCAT to analyse the raw VCF files available at GEO under accession GSE84073 (see the Supplementary Code for details and Supplementary Data 1 for the study metadata). The overall SNV-based genetic similarities between tissues and organoids are clearly grouped according to their respective patient of origin, as can be seen in Figure 1A. We also investigated if this holds true for SNV profile subsets containing only coding and missense variants. The original VCF files were thus annotated using snpEff24, followed by creation, reading and sub-setting of SNV profiles. Figure 1B shows the pairwise comparisons of these variant subsets, indicating that groupings based on genetic similarities of missense variants also separate the dataset in a per-patient manner. Comparisons with COSMIC liver variants were also performed, although the relatively tiny number of variants (no more than 23 at most) make these comparisons less informative and statistically relevant. This data covers upwards of hundreds of thousands of overlapping variants for each non-COSMIC pairwise comparison (Table 1).
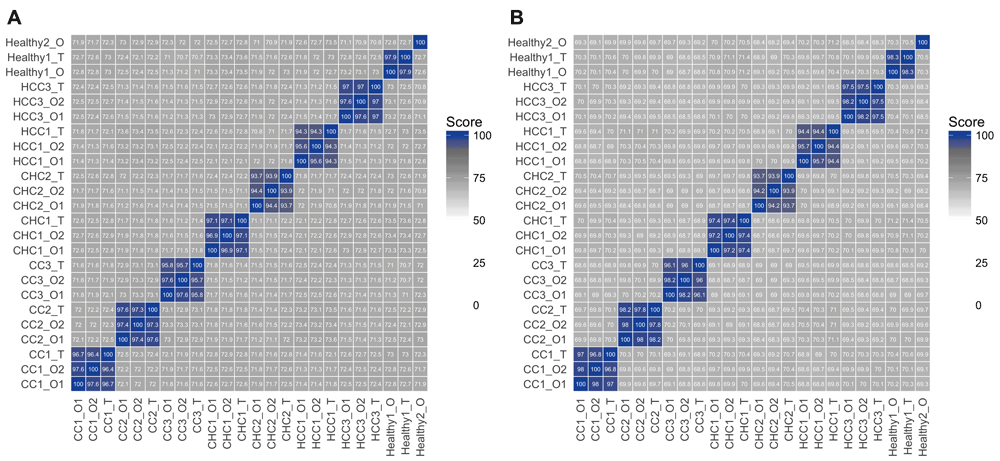
Pairwise comparisons of all WES SNV profiles, showing the genetic similarity of all individual samples for either no variant sub-setting (A) or sub-setting for coding variants only (B). The colour gradient is defined for ranges of the similarity score: scores between 0 and 50 are shown as white, scores between 50 and 90 as a white-to-grey gradient and, finally, a grey-to-blue gradient for 90 to 100. Samples are named according to their type: original tissues (T), established organoids (O1) and long-term cultured organoids (O2). These figures were created using the plot_heatmap seqCAT function.
We sought to investigate the genetic stability of the organoids both in terms of their transition from primary tissue to organoid culture, as well as long-term culturing. Figure 2A shows a boxplot of genetic similarities for both of these comparisons, indicating that the long-term cultures seem to be more genetically similar than the transition from tissue to organoid at the SNV-level. This conclusion is not statistically significant, however, with p-values of 0.36 and 0.41 for all and subset variants, respectively (Supplementary Code). A larger cohort may thus be needed to fully explore the difference between tissue-to-organoid and long-term-culturing stability. The overall high genetic similarities of all the organoids are clear, however: the lowest median similarity score across all patients and all variants is 93.9 (patient CHC2), while reaching as high as 97.9 (healthy patient 1); see Table 1. The similarity scores across coding and non-subset profiles are roughly equivalent.
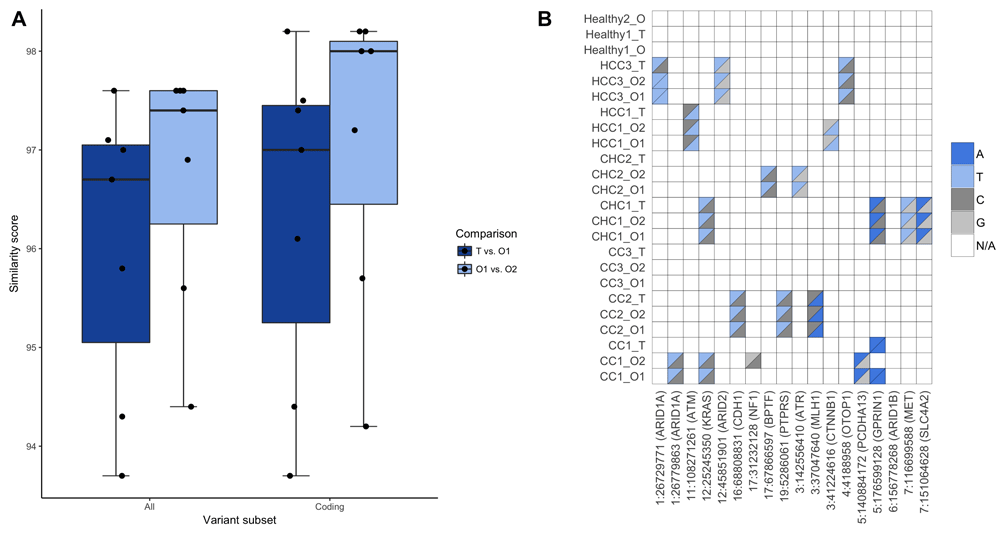
(A) Comparisons of genetic similarities between original tissue, derived organoids and long-term cultured organoids. Results are shown for both non-subset variant comparisons and for subsets including coding variants only. The differences between T vs. O1 and O1 vs. O2 for each subset are not statistically significant (α = 0.01). (B) Analysis of previously known liver cancer SNVs as listed in the original publication, where the genotype of each individual variant is visualised by different colours. White squares indicate that no confident variant was called for that position in that particular sample. This figure was created using the plot_variant_list seqCAT function.
The original publication27 lists a number of previously known liver cancer variants (Supplementary Data 2), which we analysed with seqCAT. This analysis reveals that some of the known variants are present in the organoids but absent in their corresponding tissue (Figure 2B). SeqCAT indicates that these specific variants would need to be investigated further, which the original authors have done in most cases. However, it revealed that the GPRIN1 variant is present in the CC1 samples, even though it is not listed in the literature-based variant list of the original publication (nor the COSMIC database). This is likely due to how seqCAT uses pre-defined variant lists, \textit{i.e.} by looking for all known variants in all samples.
Annotations with snpEff include variant impacts, which are the predicted effects on protein functions and range from HIGH, MODERATE, LOW through MODIFIER, in decreasing order of importance. An example of a HIGH impact is a variant leading to protein truncation, while a MODIFIER variants is predicted to a little to no effect on their resulting protein (such as intronic variants). SeqCAT can summarise and visualise these impacts across profile comparisons. Figure 3 shows the impact distributions of matching and mismatching variants for an aggregation of all comparisons between samples in the tissue-to-organoid transition as well as through the long-term culturing process.
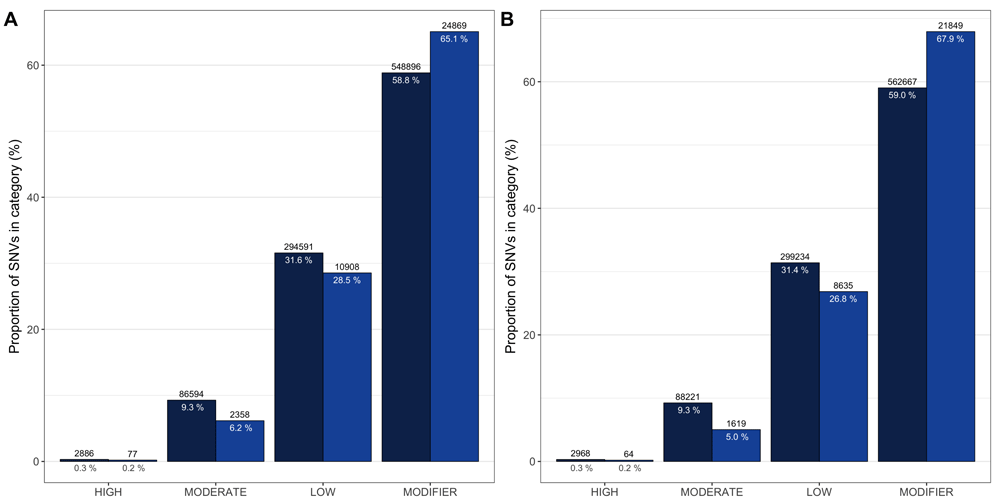
Distribution of variant impacts for the an aggregate of all pairwise comparisons between tissue and early organoid cultures (A), and early versus late organoid cultures (B). Matching variants (i.e. variants with identical genotypes for both samples being compared) are dark blue, while mismatching variants are a lighter shade of blue. These figures were created using the plot_impacts seqCAT function.
In order to investigate if any of these mismatching variants are biologically relevant, we performed GO (Gene Ontology) and KEGG (Kyoto Encyclopedia of Genes and Genomes) enrichment using DAVID28 on genes affected by mismatching variants in the HIGH and MODERATE impact categories. While no terms were significantly enriched for the tissue-to-organoid transition (α = 0.01), three olfactory-related terms and one related to protein de-ubiquitination were significantly enriched for long-term culturing comparisons (see Supplementary Data 3 and Supplementary Data 4).
In summary, these results corroborate the original authors’ conclusion that the organoids are genetically stable in vitro models of liver cancer and demonstrate how seqCAT can be used to analyse genetic variation in HTS data.
The Broutier dataset contains not only WES data but also RNA-seq data on the same samples, enabling comparison of RNA-seq data to the already performed WES analyses. We thus downloaded the publicly available raw FASTQ files, performed read alignment with the 2-pass mode of STAR29, variant calling using GATK30 and annotation using snpEff24, as previously described21. We subsequently used seqCAT to create SNV profiles for each RNA-seq sample and performed pairwise comparisons across all WES and RNA-seq SNV profiles. This resulted in a grouping with high similarities between WES and RNA-seq samples for the same patient (Figure 4).
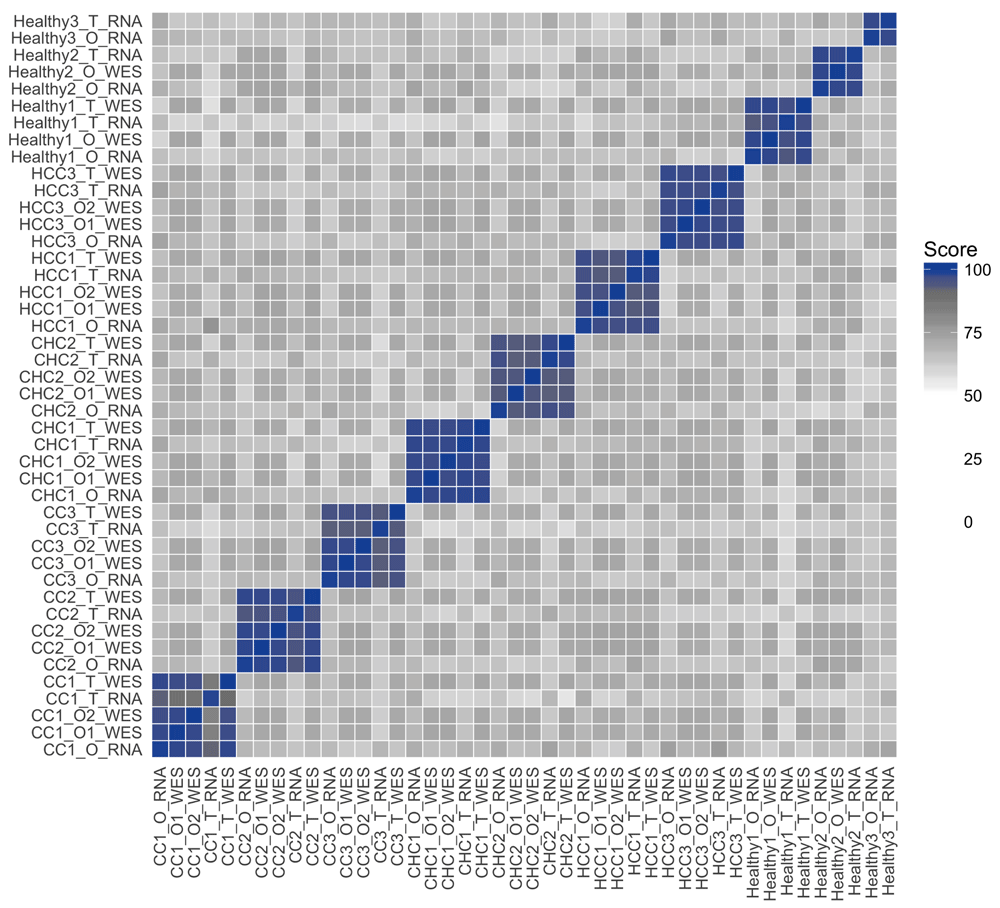
The colour gradient is the same one used for Figure 1: scores between 0 and 50 are white, scores between 50 and 90 are shown with a white-to-grey gradient, and a grey-to-blue gradient for scores between 90 and 100. This figure was created using the plot_heatmap seqCAT function.
There are several previously published studies that show discrepancy between DNA and RNA variants with varying extent and proposed causes31,32. In order to quantify the differences between DNA- and RNA-based variants in the organoid dataset, the median concordance for all same-sample comparisons was calculated to be 97.5%; the concordance was used in lieu of the similarity score in order to increase comparability with previously published results. This was also performed for sample type-specific comparisons, where the concordance for tissue versus tissue comparisons was 96.5% and 97.7% for organoid versus organoid. Per-patient (e.g. CC1 vs. CC1) calculations were also performed, shown in Table 2. The minimum per-patient concordance was 94.8% and the maximum 98.9%, while the minimum for any individual comparison was 81.1% and a maximum of 99.0% (see the Supplementary Code for the calculations). The minimum value of 81.1% (tissue versus tissue for patient CC1) is the only DNA/RNA comparison with a concordance lower than 90%. These concordances are generally higher than the 80 to 90% that have previously been shown32, but still highlight a difference between DNA and RNA variants. While different explanations for this discrepancy has previously been suggested (such as RNA editing), a deeper investigation of these is outside the scope of this paper.
| Patient | Median concordance | Median overlaps |
|---|---|---|
| CC1 | 96.9% | 3744 |
| CC2 | 98.9% | 609 |
| CC3 | 98.2% | 718 |
| CHC1 | 97.9% | 3164 |
| CHC2 | 95.1% | 920 |
| HCC1 | 96.5% | 1872 |
| HCC3 | 97.3% | 745 |
| Healthy1 | 94.8% | 1606 |
| Healthy2 | 98.7% | 1367 |
In summary, results from seqCAT demonstrate an overall high level of concordance between DNA and RNA variant calls, but highlight that there is some variation between sample types and patients.
HTS experiments are becoming increasingly more common and the need for simple and powerful bioinformatic software is as great as ever. Analyses of genetic variation through e.g. SNVs represents a common endeavour for many scientific studies, but the methods and data analysis pipelines used vary. In this study we present seqCAT, an easy-to-use and well-documented Bioconductor23 R-package that performs variant analyses of HTS data. The capabilities of seqCAT include the creation of SNV profiles, comparisons of global genetic similarities for all variants common between samples and analyses of single variants or genes of special interest. While the seqCAT package itself is new, the underlying theory and general methodology have previously been used for investigations into cell line authenticity21 and genetic heterogeneity in public cell line datasets22.
SeqCAT may be used to analyse both novel sequencing data as well as publicly available data in repositories (such as the GEO)1, but may also be utilised to define genetic profiles for any sample of interest. Such profiles are of great interest for researchers using model systems (such as cell lines or organoids), as it allows for a clear definition of the genetic background of the model itself. This could then be referred back to at a later time, to make sure that genetic drift (that obscure interpretation of biological results) has not occurred. SeqCAT is both easy to install and to use, and includes in-depth documentation on its functionality and underlying theory.
In the present study, we have used seqCAT to analyse a publicly available dataset containing WES and RNA-seq data from organoid cultures and their tissues-of-origin27. The global analysis of WES SNVs demonstrate the overall high genetic similarities between the organoids and their respective tissues, with equivalent results for comparisons covering all variants or only missense variants. The seqCAT-analysis of known variants indicate that a GPRIN1 variant is present for the CC1 patient; this variant is only listed as present in a CHC-type patient in the original study. The SNV-based results presented herein corroborate the original authors’ conclusions that organoids are genetically stable over time, but the higher level of genetic similarity between early and long-term cultured organoids as compared to the tissue-to-organoid transition is statistically non-significant.
The analyses of genes affected by mismatching HIGH and MODERATE impact variants show that none of the differences between tissue and initial organoid cultures are significantly enriched for specific biological functions, indicating that these differences likely are random. The transition from primary tissue to organoid can thus be viewed as a stable transition, especially given the high overall similarity previously discussed. The long-term culturing results do, however, present four significantly enriched terms. Three of these are related to ectopic expression of olfactory receptors, which have previously been shown to be present in both healthy and cancerous tissues33,34. The single GO-term related to protein de-ubiquitination may be important for studies investigating ubiquitination in liver cancer. Both of these points should thus be accounted for when performing a study with these organoids. The overall results yielded by the seqCAT-analyses corroborate the conclusions from the original study, i.e. that these organoids are genetically stable and may be suitable models for studying liver cancer.
There have been several studies comparing variant calls from DNA and RNA of the same samples, but they have come to differing conclusions as to both the extent and causes of the DNA/RNA discrepancies. Li et al. performed both DNA/RNA-seq across 27 individuals in addition to analyses of protein expression using mass spectrometry, where peptides corresponding to variants found in both DNA and RNA were present31. They argue that their results indicate biological significance of RNA variants, given that they are translated to proteins, and that the differences between DNA and RNA variants can be biologically meaningful. Indeed, there have been several studies analysing RNA-seq variants that yielded novel biological insights, demonstrating the utility of such endeavours35–39. A study by Guo et al. analysed DNA/RNA-seq data for 10 breast cancer patients from the TCGA and calculated DNA/RNA concordances to range between from 80 to 90%32. They argue that these differences are mostly technical rather than biological.
The results of the present study indicate that the extent of DNA/RNA differences may not be as large as previously shown: the median concordance for DNA/RNA pairs was 97.5% overall, with a range of 90 to 99% (plus a single comparison with 81.1%), while Guo et al. reported a range of 80 to 90% concordance. Both studies thus find a discrepancy between DNA- and RNA-based variant calls, but disagree on its extent. The RNA-seq pipeline utilised in this study is based on the current best practices of GATK, which uses the STAR software for read alignment that has proven to be highly accurate for RNA-seq data29,40. The latest assembly of the human genome (GRCh38) was also used, as the choice of assembly has been highlighted as an important parameter that can yield higher accuracy32. Guo et al. used an earlier assembly from 2009 (GRch37), which might partly explain the discrepancy between the results. The choice of sequencing platform and differences in mutational profiles of breast and liver cancer could also be affect the comparisons. While technical issues will always exist even for DNA/DNA or RNA/RNA comparisons, the results of the present study may represent a closer estimate of the biological relevance of DNA/RNA differences first noted by Li et al.
It is clear is that there is a discrepancy between DNA- and RNA-based variant calls, but the exact extent of this difference remains to be determined, as well as whether it is a consequence of technical artefacts or biological variation. A full evaluation of these matters likely require a larger study than what has previously been attempted, including using the latest technologies as well as protein-level validation. The analyses performed herein demonstrate how seqCAT may be utilised as a part of such an endeavour.
The seqCAT Bioconductor R-package provides an effective and easy-to-use toolkit for analysing HTS variant data, enabling researchers to investigate genetic differences and potential variation within and between their samples or publicly available data from other laboratories. Little R expertise is required to use seqCAT, and its use is extensively documented. We have used seqCAT to analyse genetic variation in a publicly available dataset of liver cancer organoids, corroborating the conclusions drawn by its original authors, as well as demonstrate high levels of DNA/RNA SNV concordance in this dataset. These results serve as a case study in how to utilise the capabilities of seqCAT, which make it a valuable and intuitive tool for a wide range of researchers.
Software is available from: https://bioconductor.org/packages/release/bioc/html/seqCAT.html
Source code available from: https://github.com/fasterius/seqCAT
Archived source code as at time of publication: https://doi.org/10.5281/zenodo.266914341
Software license: MIT
The data used in this article is publicly available at the GEO through the accession number GSE84073.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)