Keywords
gut microbiota, hygiene hypothesis, factors influencing gut microbiota, 16S rRNA sequencing
gut microbiota, hygiene hypothesis, factors influencing gut microbiota, 16S rRNA sequencing
The relationship between human gut microbiota and certain diseases has become increasingly apparent1,2. The main contributing factors are delivery mode3–5, age6,7, antibiotic treatment4,6, diet, and the living environment. They affect gut microbiota to different degrees. Among these factors, diet is the most widely studied one, as it modulates the composition and then changes the function of the microbial community in humans8–15. The microbiota of malnourished children was shown to impair growth and cause metabolic abnormalities in the brain and other organs of recipient gnotobiotic mice16,17. By altering the gut microbiota, diet also contributes to other chronic illnesses, such as obesity1,2, cardiovascular diseases18, and autism19. The human gut microbiota responds rapidly to dietary changes8,20, even though long-term dietary habits remain the dominant force in determining the composition of an individual’s gut microbiota10,15,21,22.
Following what is known as the hygiene hypothesis, an overly clean modern life leads to immune dysfunction and more allergic diseases23,24. Many studies have shown large differences in the human gut microbiome between populations in rural areas across the world and those in Europe or America7,9,25. Our early research suggested that the cleanness of a living environment substantially altered the composition of gut microbiota in mice26. After analyzing the elements in bedding material, it was first proposed that microbes might play a key role in changing the composition of mouse gut microbiota27. However, a follow-up study showed that adding microbes to the bedding had a limited effect on the composition of the dominant genera28. Thus, the unsanitary environment factors that act on gut microbial communities remain to be identified.
Unsanitary environments are rich in surface soil. This hosts a large number of microbes and is rich in nutrients. At present, it is unclear what effect the soil in our environment exerts on the composition of our gut microbiota, to what extent it causes such alterations, and what the time-scale of any changes may be. To understand these interrelated issues, we raised C57 mice on four different beddings. Groups were designed to explore the effect of sterile and unsterile soil, the extent by which soil compared with normal diet or high-fat diet (formula shown in Supplementary Table 1), as well as the timing of exposure to soil (before birth or after weaning) on the gut bacterial community (Supplementary Figure 1).
We analyzed the bacterial community composition using MiSeq sequencing targeted to the 16S rRNA gene in up to 180 fecal samples (Supplementary Table 2). For each sample, we PCR-amplified the V4 hypervariable region using the 515F-907R primer set29. The sequencing data set consisted of 10,937,871 high-quality, classifiable 16S rRNA gene sequences, with at least 45,816 sequences per sample (Supplementary Table 2).
On the basis of the obtained sequences, we discovered that sterile and unsterile soil significantly altered gut bacterial community composition (Figure 1 and Supplementary Table 3) in a diet-dependent manner. Mice fed a normal diet and living on sterile soil after weaning exhibited significantly more Bacteroidetes (P < 0.001) and fewer Firmicutes (P < 0.001) (Figure 1A and Supplementary Table 3a). Unsterile soil added before birth also significantly increased the number of Bacteroidetes (P = 0.032). Instead, mice fed a high-fat diet and raised on bedding with unsterile soil added before birth or after weaning showed significantly more Actinobacteria (P < 0.001) (Figure 1B and Supplementary Table 3b).
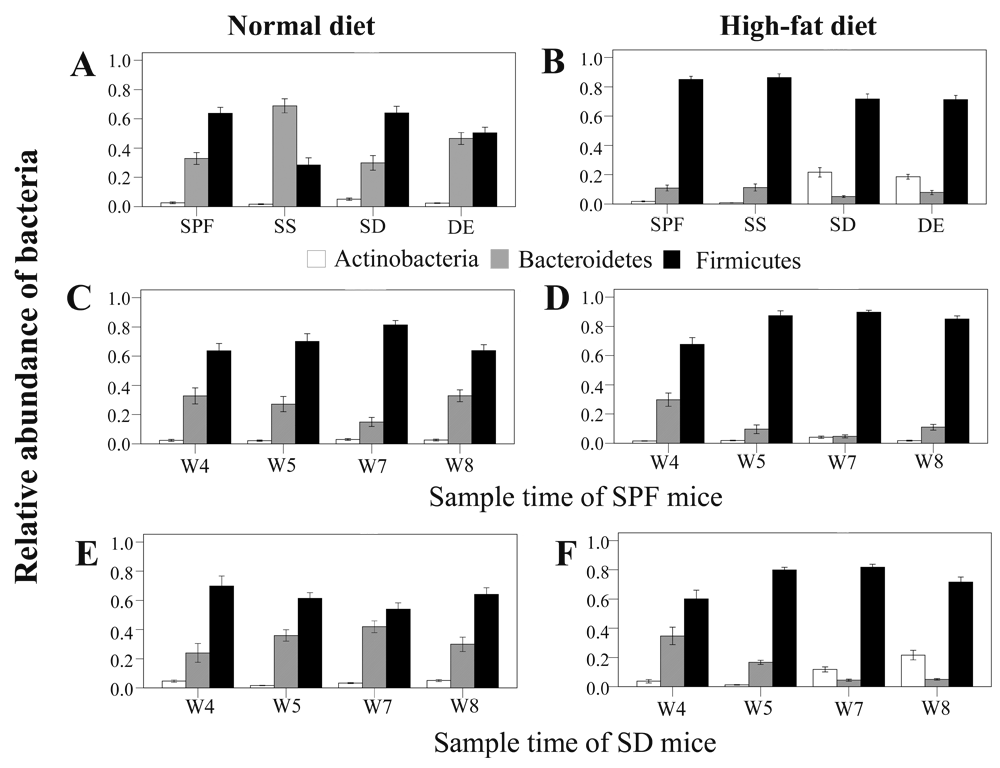
Values from all available fecal samples were averaged (n = 8, 9, or 10 per treatment). (A,B) Relative abundance of bacteria on the eighth week in fecal samples of mice raised on specific-pathogen-free grade (SPF) sterile bedding (control), sterile bedding with sterile soil added after weaning (SS), sterile bedding with dirty unsterile soil added after weaning (SD), and sterile bedding with unsterile soil added prior to birth (DE) fed (A) a normal diet (ND) or (B) a high-fat (HF) diet. (C–F) Relative bacterial abundance in samples collected on the fourth (W4), fifth (W5), seventh (W7), and eighth (W8) week from mice of the (C) SPF-ND group, (D) SPF-HF group, (E) SD-ND group, and (F) SD-HF group.
Microbes in the environment were reported to affect the colonization of the intestinal microflora in newborns30. Here, we found that microbes and soil in the bedding could change the composition of gut microbiota after mice were weaned. When mice were fed a normal diet and lived on bedding with unsterile soil, they consistently showed more Bacteroidetes (P < 0.001) and fewer Firmicutes (P < 0.001) (Figure 1E and Supplementary Table 4a) during the first 3 weeks. After the third week, the abundance of these two phyla returned to the level from the previous week. When fed a high-fat diet and provided with bedding containing unsterile soil, mice showed an increased abundance of Actinobacteria on the third week of treatment (P = 0.030), becoming highest on the fourth week (P < 0.001) (Figure 1F and Supplementary Table 4b). In summary, we observed that the gut microbiome could respond rapidly to alterations in the living environment and diet, potentially facilitating adaptation to a variety of lifestyles.
We further discovered that unsterile soil could increase microbial diversity, particularly for mice on a normal diet, whether it was added before birth or after weaning (Figure 2, Supplementary Figure 3, and Supplementary Table 5, Supplementary Table 6). By comparison, microbial diversity was unaffected by sterile soil in mice fed a normal diet and decreased in those on a high-fat diet (Supplementary Figure 3 and Supplementary Table 5). Earlier, we found that merely adding environmental microbes to the bedding could increase diversity but had little effect on the composition of dominant bacteria in mice gut microbiota28. Hence, microbes appear to contribute mainly to microbial diversity, whereas soil may alter microbial community structure.
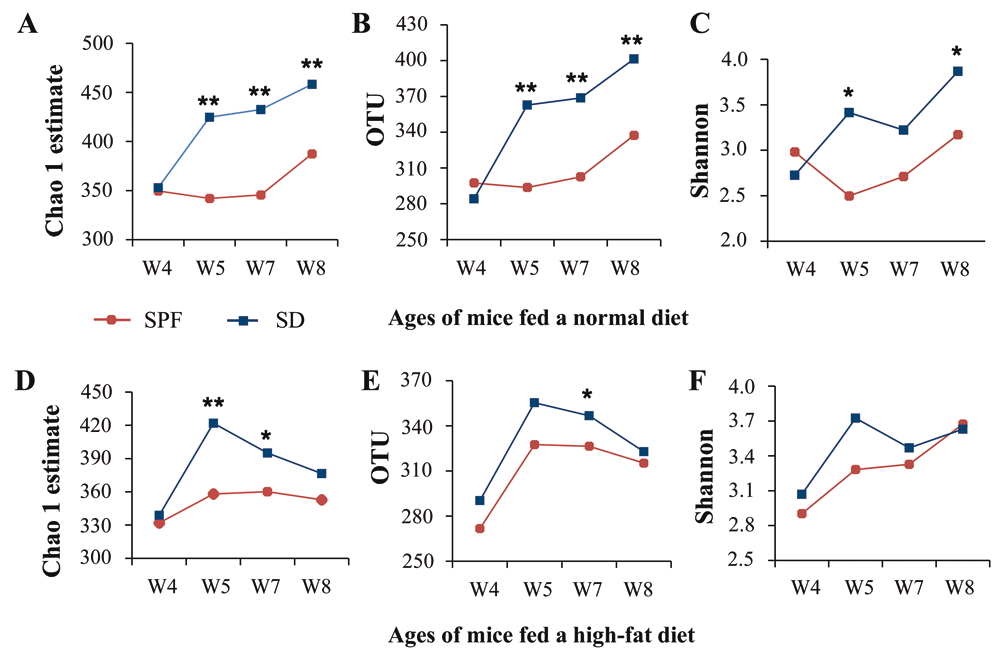
(A–C) SPF and SD groups fed a normal diet: (A) operational taxonomic units (OTUs), (B) Chao 1 estimates, and (C) Shannon index. (D–F) SPF and SD groups fed a high-fat diet: (D) OTUs, (E) Chao 1 estimates, and (F) Shannon index. Values from all available fecal samples were averaged (n = 8–10; *P < 0.05, **P < 0.01, based on a two-tailed least significant difference test).
An epidemiological survey has shown that immune system diseases, such as asthma and wheeze, display a skewed sex bias towards males31. Here, we found that abundance of the phyla Bacteroidetes and Firmicutes was related to gender, except in mice with sterile bedding with unsterile soil added prior to birth (DE group) exposed to unsterile soil bedding from birth (Supplementary Figure 4, Supplementary Figure 5 and Supplementary Table 7). Bacteroidetes were more abundant in males than in females of the mice raised on specific-pathogen-free bedding (SPF group) fed a normal diet (P = 0.019). By contrast, Firmicutes were more abundant in males than in females in the mice raised on sterile grade murine bedding with dirty unsterile soil (6:11 (w/w)) added after weaning (SD group) on a high-fat diet (P = 0.042) (Supplementary Figure 5 and Supplementary Table 7).
Random Forests is a supervised machine-learning technique that can classify a sample by estimating the importance of OTUs at species level according to their relative abundance and sample probability32. We found that distinct community signatures existed between any two treatments (Supplementary Table 8–Supplementary Table 10). For mice on a high-fat diet, there were 79 predictive species-level OTUs between the (SPF) and DE groups (baseline error = 0.44, cross-validation error = 0 ± 0), of which 63 were overrepresented in the DE group. Moreover, out of these 63, 25 and 20 were assigned to the classes Actinobacteria and Bacilli, respectively (Supplementary Table 8b). A comparison between SPF and SD groups gave similar results (Supplementary Table 8a); however, the decrease in beneficial bacteria was greater in the mice raised on sterile grade murine bedding with sterile soil (6:11 (w/w)) added after weaning (SS group) than in the SPF group (Supplementary Table 8c).
We further analyzed differences in the composition of gut bacterial communities between any two groups of mice on a normal diet (Supplementary Table 9). Compared to the SPF group, DE and SD groups presented more Actinobacteria and Bacilli, whereas the SS group showed more Bacteroidia (Supplementary Table 9). Notably, bacteria of the Bacteroidales S24–7 family were more prevalent among SPF mice fed a high-fat diet than in those fed a normal diet. Instead, the latter had more bacteria of the families Lachnospiraceae and Ruminococcaceae (Supplementary Table 10a). The DE group fed a high-fat diet showed more Actinobacteria and Bacilli than animals fed a normal diet. On the contrary, the latter had more Bacteroidales S24–7 and Lachnospiraceae (Supplementary Table 10c). Thus, unsterile/sterile soil bedding affected the structure of mice gut microbial communities in a diet-dependent manner. The effect varied depending on whether i) mice were raised on bedding with soil added before birth or after weaning, ii) the bedding contained sterile or unsterile soil, and iii) mice were fed a normal or high-fat diet.
We used UniFrac distances to analyze the 16S rRNA datasets and measure similarities among microbial communities. The distance between mice exposed to two different living environments and fed a high-fat diet was similar to that between mice using the same bedding, and fed either a normal or a high-fat diet (Figure 3 and Supplementary Table 11). The distance between SPF groups fed a normal or a high-fat diet did not differ significantly from that between SPF and SS, and between SD and DE mice fed a high-fat diet. Indeed, these groups were among the least distant. Distances between mice maintained on different beddings and a high-fat diet were greater than those between SPF groups fed different diets. Thus, soil appears to influence the composition of the gut microbial community to the same extent as diet. In addition, a change in living environment also had an important effect on gut microbiota. When fed a normal diet, microbiota in the gut of SPF and DE mice were more similar than those of other groups (Figure 3 and Table 11).
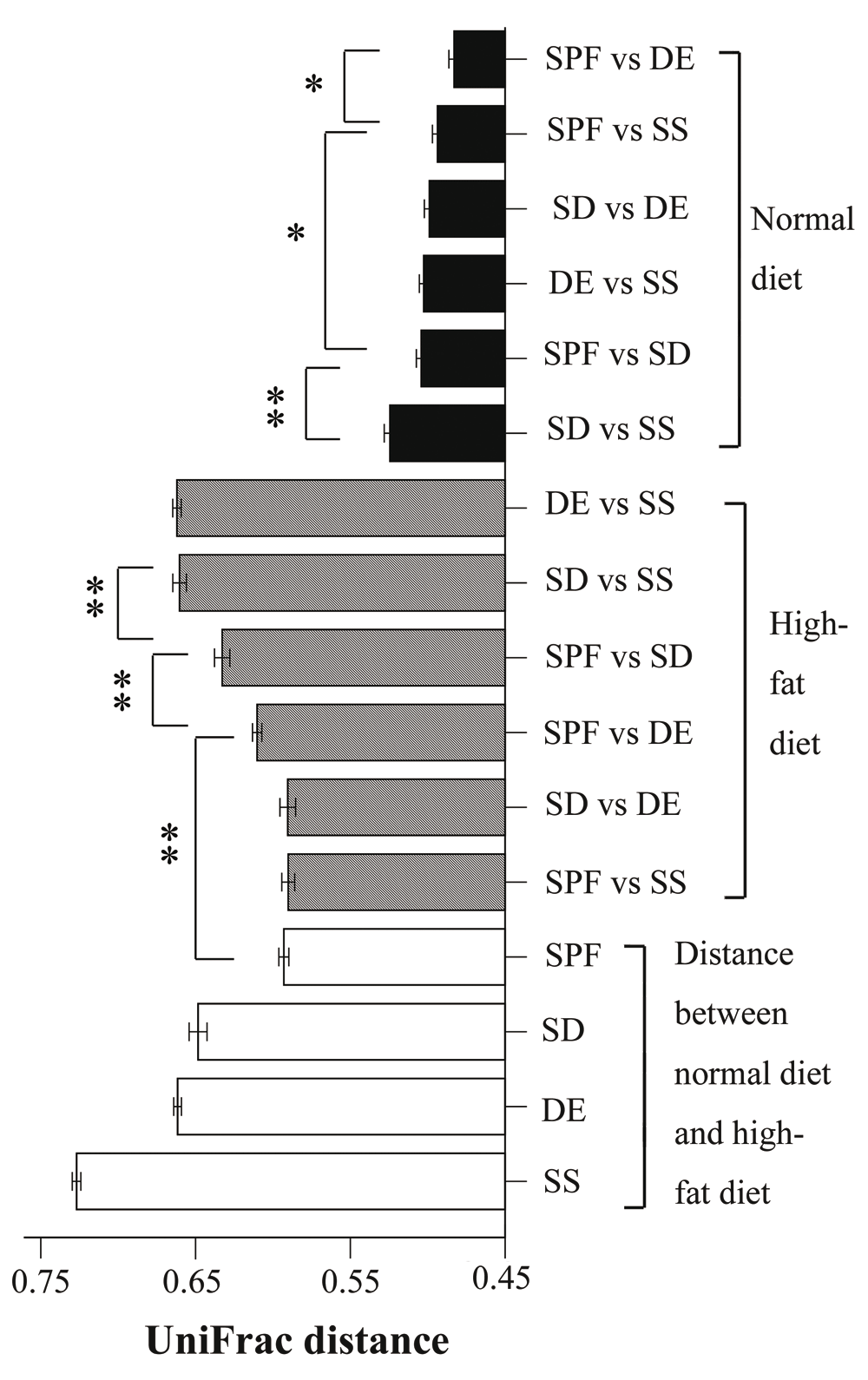
For mice fed a normal diet, the shortest UniFrac distance was between specific-pathogen-free (SPF) and sterile bedding with unsterile soil added prior to birth (DE) groups, and the longest one was between sterile bedding with dirty unsterile soil added after weaning (SD) and sterile bedding with sterile soil added after weaning (SS) groups; comparisons between SPF and SD, and between DE and SS showed no significant differences. For mice fed a high-fat diet, the shortest distances were SPF–SS and SD–DE, with no significant difference between them; the longest distances were SD–SS and DE–SS; SPF–SD was significantly longer than SPF-DE (n = 8–10; *P < 0.05, **P < 0.01, based on a two-tailed least significant difference test).
Unsupervised clustering using PCoA of UniFrac distance matrices confirmed that living environments and diets explained the variation in the data set (Figure 4 and Supplementary Figure 6, Supplementary Figure 7). When fed the same diet, the gut microbial communities of mice from one living environment clustered apart from those of mice subjected to other treatments (Figure 4A, B). For two different living environments, the two high-fat diet groups showed a similar transfer distance as mice maintained on the same bedding but fed two different diets (Figure 4C, D and Supplementary Figure 6). For the SPF group fed a normal diet, almost no differences were observed among fecal samples collected at four different time points (Supplementary Figure 7A). These results confirm that the living environment exerts a great influence on the composition of gut microbiota. It should be noted that we did not find significant clustering with respect to cage or gender.
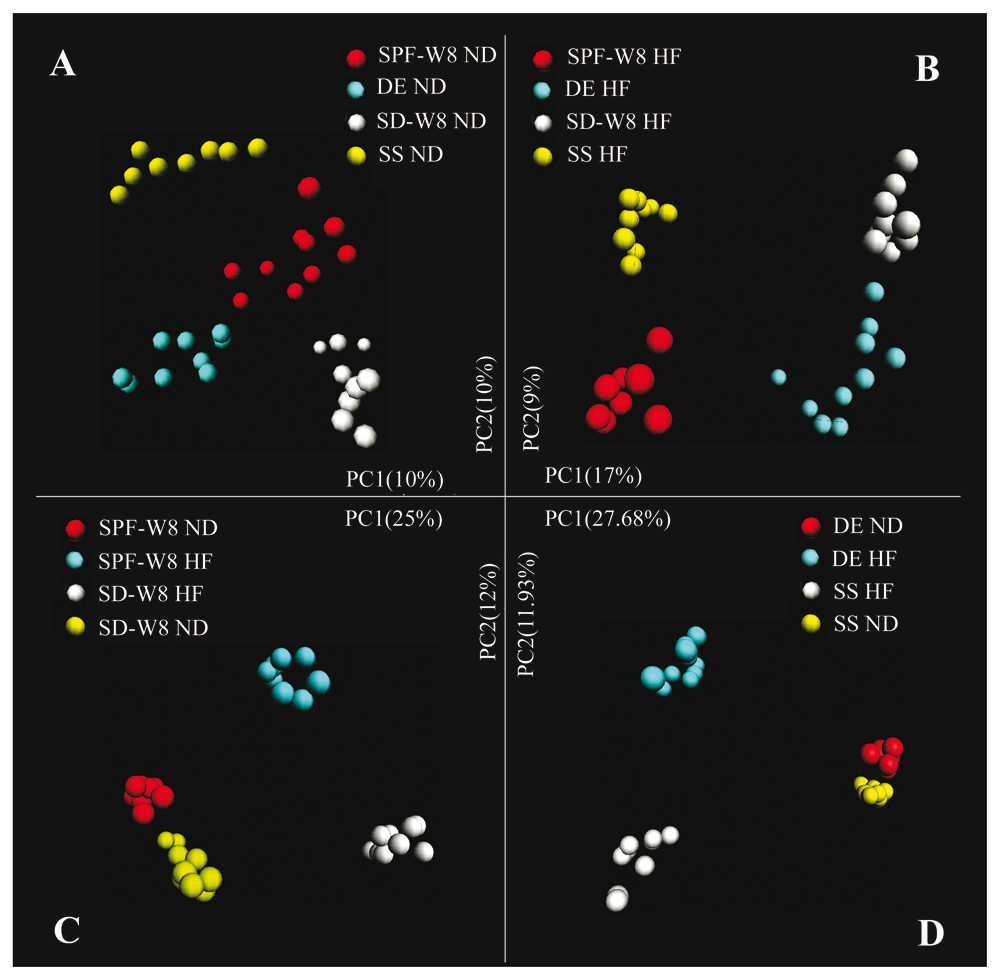
Analysis was based on the Illumina bacterial 16S rRNA gene data set (V4 region). Mice from the four groups were fed a (A) normal or (B) high-fat diet. (C) Mice in the sterile bedding with dirty unsterile soil added after weaning (SD) and specific-pathogen-free (SPF) groups were fed a normal or high-fat diet. (D) Mice of the sterile bedding with sterile soil added after weaning (SS) and sterile bedding with unsterile soil added prior to birth (DE) groups were fed a normal or high-fat diet.
In the present study, we identified differences in gut microbiota between mice living on sterile or unsterile soil and fed a normal or a high-fat diet. Sterile and unsterile soil substantially altered the composition of gut microbiota, with unsterile soil also increasing bacterial diversity. In addition, we demonstrated that unsterile soil added before birth or after weaning altered the gut microbiome, irrespective of whether mice were fed a normal or a high-fat diet. Hence, we suggest that soil is one of the key factors influencing gut microbiota and that its effect is comparable to that exerted by diet.
Mice have the habit of coprophagy and fur combing, through which they may swallow some soil. As a result of agriculture and animal husbandry, our ancestors were also in a close and daily contact with soil. This practice is still common among infants (age 12–14 months), who crawl on grounds rich in soil and animal feces. During this period, they use their mouths to feel the world around them and experience a rapid development of the immune and nervous systems. Our results may explain in part some epidemic diseases occurring in modern life.
We show that sterile soil present in the surrounding environment has a role in shaping the composition and structure of gut microbiota. At birth, the gut is sterile and ambient microbes play a key role in shaping the bacterial community3–6,30,33. Our early research showed that microbes in the environment had a limited capacity to influence the composition and structure of dominant bacterial communities in adult mice28. Here, we show that sterile soil in the bedding significantly altered the composition and structure of gut microbial communities in weaned mice, but had little effect on their diversity and richness. By contrast, unsterile soil changed microbiota composition and structure, as well as its diversity and richness. Together, these studies indicate that a healthy gut microbiota benefits from a close contact with soil and the microbes in it. It is possible that unsterile soil promotes the growth of certain bacteria, while inhibiting others, especially in mice fed a high-fat diet.
The most obvious effect of unsterile soil on the intestinal microflora was the predominance of the phylum Actinobacteria in mice fed a high-fat diet. Vatanen et al.34 studied the development of gut microbiota in infants from birth until the age of 3 years and found that early-onset autoimmune diseases were common in Finland and Estonia but were less prevalent in Russia. Their results showed that Actinobacteria were more abundant among infants under 12 months in Russia than in Finland and Estonia; however, there was no difference after the age of 1 year34. This might be explained by small infants having a diet rich in protein and fat, and those in Russia being in closer contact with unsterile soil.
The results also indicate how the timing of exposure to unsterile soil influences the formation of bacterial communities. Some studies reported that early environmental exposure exerted a sustained influence on the acquired gut microbiota, and that the early microbiota shaped the later one35–37. Our results reveal that microbiota differed depending on whether mice were exposed to unsterile soil early or after weaning. It remains to be determined whether the difference between gut microbiota of mice that were exposed to soil before birth or after weaning influences the animals’ health.
Our synergetic assessment of host soil and diet provides additional insight into the effect of diet on gut microbiota, and the possible effects on colorectal cancer and immune system diseases. We speculate that differences in bacterial flora mirror a different metabolite composition. Epidemiological studies indicate that red and processed meat intake is associated with an increased risk of colorectal cancer38,39 and diseases of the immune system40. However, in pastoral areas, such as Tibet, where the local diet is rich in beef and mutton, mortality from cancer and childhood asthma is the lowest in China41,42. Compared with a modern western lifestyle, the residents of Tibet are in close daily contact with soil. To this end, unsterile soil may alter microbiota composition and structure, increasing its diversity and richness, and affecting the metabolic processing of red meats. This, in turn, may enhance human immunity and resistance to chronic diseases; however, this mechanism needs to be confirmed via further research.
Our results provide further evidence in support of the hygiene hypothesis. In developed countries, human living environments and lifestyle have changed immensely during the last few decades. People spend ~92% of their time indoors43 and there is very little soil in their living environment. At the same time, many microbes appear to have been lost from the modern human body7,9,25, while the burden of chronic inflammatory diseases, including atopic diseases, has increased dramatically. Our results suggest that an important cause of these diseases is represented by the lack of contact with soil and soil microorganisms.
The cleanness of our living environment and diet could be readily modified. This approach may provide a new simple route to intervene on the human gut microbial community, as well as design specific diets aimed at disease prevention. A few open questions nevertheless remain. Further research will determine how the soil from the host environment alters human gut microbiota, whether through swallowing, body mucosae, or by other means. Similarly, it remains to be investigated whether different types of soil or soil microbes have different effects on intestinal microecology.
We purchased 6-week-old male (n=1) and female (n=3) C57 mice from B & K Universal (Shanghai, China) for breeding. One male was mated with three females. After the females became pregnant, two of them were shifted to a separate cage. The newborn mice were weaned and when they were 4 weeks old they were moved to separate cages.
We selected 8 males and 24 females from among the weaned mice to carry out further experiments. Next, two males and six females were selected from different cages and transferred to room number 2. The cages were furnished with sterile grade murine bedding and unsterile soil (6:11 w/w bedding to soil). The remaining 6 males and 18 females were raised in room number 1, on sterile grade bedding. The bedding was changed once a week. The soil used in the experiments was from the top 10 cm of a farm ground with abundant goats, hens, and ducks.
When mice were 6 weeks old, they were bred with 1 male per 3 females. In room number 1, 30 males and 30 females, aged 23–37 days (weight, 8.6–21.4 g) the fourth week group, W4), were selected for experiments. They were randomly separated into three groups: the SPF group, raised on specific-pathogen-free bedding; the SD group, raised on sterile bedding with unsterile soil (6:11 (w/w)) added after weaning; and the SS group, raised on sterile bedding with sterile soil (6:11 (w/w)) after weaning. The SPF group served as the control. The randomization procedure was as follows. Considering gender matching, more than 80 mice were used in this experiment, and it was very difficult to breed so many mice in the same time. When we had about 20 mice with an age difference within about 10 days, we divided them into two groups, one fed with normal diet and the other with a high-fat diet. These 20 mice had been treated with same bedding. The other groups of 20 mice were treated with the second or third of type bedding. The breeding mice were uniformly distributed and randomly grouped. The SD group was transferred to room number 2 after weaning. Next, 10 males and 10 females, who were born in room number 2, aged 23–33 days (W4) were selected for experiments and raised on the same kind of bedding as before weaning (group DE). For all experiments, two or three mice were kept in a cage and the bedding was changed once a week.
For each group, 10 mice (five males and five females) were fed the same sterile grade commercial normal pellet diet as before weaning, while 10 mice (five males and five females) were fed a sterile grade commercial high-fat pellet diet after weaning (Supplementary Table 1). All animals were fed the same sterile distilled water. They were allowed free access to water and food. All animals were housed in two specific-pathogen-free animal rooms with a 12-h light/12-h dark cycle at 24°C ± 2°C and humidity of 40% ± 5%.
For SPF and SD groups, the first fecal samples were collected before mice were shifted to separate cages, and then on the fifth, seventh, and eighth week (Supplementary Figure 2). For DE and SS groups, fecal samples were collected on the eighth week (Supplementary Figure 2). After collection, fecal samples were put immediately into an ice box and stored at −80°C within 8 h.
Animal experiments were performed in strict accordance with the guidelines of the Animal Research Ethics Board of Southeast University. All experiments were approved by the Animal Care Research Advisory Committee of Southeast University and the National Institute of Biological Sciences (approval number: 2014063009). All efforts were made to alleviate the suffering of animals.
Specifically, mouse health was monitored every other day and body weight measurements were performed every week. Mouse health was assessed by observing changes in body weight or fecal shape, and external physical appearance. The animals would be euthanized to minimize pain and distress if they became severely ill over the experimental period, as when they showed one or more symptoms, such as 15–20% weight loss, diarrhea, loss of hair quality, pain (arched back and curled up posture), or listlessness for more than 1 week.
Euthanasia was performed as follows. A 1% (w/v) dose of sodium pentobarbital (50 mg/kg) was administered through an intraperitoneal injection using a hypodermic needle. When mice lost consciousness, they were killed by neck dislocation and death was confirmed. Two individuals were required to perform injections, one to hold the animal and the other to perform the procedure. Animals were not left unattended during the procedure. The ARRIVE reporting guidelines were followed during this study (Supplementary File 1)44.
Genomic DNA was extracted from fecal samples according to the protocol proposed by Zoetendal et al.45. Each sample (40–60 mg) was weighed and put into 15 ml 1 × PBS (pH 7.4). Samples were re-suspended completely by vigorous shaking and subsequently centrifuged at 700 × g at 4°C for 5 min. The supernatants were transferred to a 15-ml tube and centrifuged at 9000 × g at 4°C for 5 min. The pellets were transferred to a 2-ml Eppendorf tube and re-suspended in 300 μl 10× TE buffer (10 mM Tris-HCl pH 8.0, and 1 mM EDTA pH 8.0). Lysozyme (100 μl, 200 mg/ml) was added to each tube, and incubated for 1 h at 37°C. Subsequently, 50 μl 10% SDS (w/v) and 20 μl proteinase K (20 mg/ml) were added to the samples and incubated for 2 h at 50°C. Next, 100 μl 5 M NaCl was added, followed by incubation at 65°C for 10 min. The samples were mixed gently with an equal volume of Tris-Phenol (pH 8.0) for 5 min. The mixtures were centrifuged at 9000 × g for 5 min. The upper layer was transferred to a new tube. This step was repeated twice, after which chloroform was used instead of Tris-Phenol to clean the samples one more time. Finally, a 1/10 volume of 3 M sodium acetate (pH 5.2) and two volumes of 95% ethanol were added. Samples were mixed gently and centrifuged at 4°C and 9000 × g for 20 min. The precipitated genomic DNA was washed twice in 200 μl 70% ethanol. The dried DNA samples were re-suspended in 50 μl sterile double-distilled water. DNA quality was monitored by gel electrophoresis and spectrophotometry (260/280 nm). The samples were stored at −20°C.
The analysis of gut microbial communities was performed using MiSeq high-throughput sequencing of bacterial 16S ribosomal RNA (rRNA) genes. For each sample, the V4 hypervariable region was amplified using the 515F and 907R primer set, as described by Angenent et al.29. PCR was performed with 300 ng microbial community DNA as a template, using 1 μl of Trans Start Fast Pfu Taq DNA Polymerase (TransGen Biotech, Beijing, China), 0.25 mM of each dNTP, 1× Trans Start Fast Pfu buffer, 0.2 μM forward primer, and 0.2 μM reverse primer, in a total reaction volume of 50 μl. The cycling conditions were: 94°C for 3 min followed by 27 cycles of 95°C for 45 s, 50°C for 45 s, and 72°C for 45 s, with a final extension at 72°C for 5 min. Each sample was amplified in duplicate, combined, purified, and then quantified using the QuantiFluor™-ST Handheld Fluorometer with UV/Blue Channels (Promega Corporation, Fitchburg, WI, USA). Sequencing of the PCR product libraries was performed on an Illumina MiSeq platform (Illumina Inc., San Diego, CA, USA) using 2 × 250 bp chemistry. We merged the raw paired-end reads using FLASh software version 1.2.7, set to a minimum overlap of 10 bp. Other parameters were set to default settings. PCR artifacts were removed by eliminating low-quality sequences using Trimmomatic version 0.3046. Chimeric sequences were removed using Usearch version7.147. We clustered the quality-checked sequences into de novo OTUs at a 97% similarity threshold using the QIIME software version 1.8.0 software package48. The generated OTUs were classified using the RDP classifier software version 11.5 at a 70% confidence threshold for sequences longer than 200 bp49.
All samples were rarefied down to 45,816 sequences per sample to prevent bias due to sampling depth. We used QIIME to calculate α diversity indices of Chao1 estimator and Shannon diversity/richness for each sample48. We calculated the metrics of unweighted UniFrac distances between any two treatments and performed principal coordinates analysis (PCoA) through QIIME to examine dissimilarities in community composition. PCoA was used to compare groups of samples based on unweighted UniFrac distance metrics by plotting n samples in (n − 1)-dimensional space.
Random Forests analysis in R version 3.2.37 was performed with 500 trees and all default settings using 16S rRNA-based OTUs from the Illumina V4 data sets as described previously50. We used out-of-box (OOB) error to estimate the generalization error for all 16S rRNA comparisons involving all treatments. For each comparison, 100 relevant subsets of samples were extracted from the table of OTUs, and the average OOB error estimates and OTU importance estimates were calculated from subset samples. For a direct evaluation of the predictive strength of the OTUs, we compared generalization errors at various sequencing depths: the lowest observed depth of 45,816 sequences and at sequencing depths of 100, 1000, 10,000, and 40,000 reads per sample. The mean and standard deviation of the OOB error were estimated for each classification task using 100 independent rarefactions of the data. The expected ‘baseline’ error was obtained by a classifier that simply predicted the most common class label.
T- test and analysis of variance with post hoc Tukey’s test, for comparison of two or more than two groups, respectively, were performed using SPSS software, version 18.0 (SPSS, Inc., Chicago, IL, USA).
Metagenomic sequence data for each mouse are available from the Sequence Read Archive, accession number, PRJNA491246: https://identifiers.org/insdc.sra/PRJNA491246.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)