Keywords
Huntington’s disease, epigenetics, chromosome conformation signature, chromatin architecture
Huntington’s disease, epigenetics, chromosome conformation signature, chromatin architecture
This updated version 2 contains a new version of Figure 2, (replacing Figures 2 & 3 in the original version) and updated text in the Introduction as per the reviewer's request.
See the authors' detailed response to the review by Willeke M.C. van Roon
Huntington’s disease (HD) is a neurodegenerative condition characterized cellularly by the loss of neurons in the basal ganglia and clinically by uncontrolled movements, emotional problems, and loss of cognition1. HD is an autosomal dominantly inherited disorder and although prevalence rates range widely depending on geography and ethnicity, it is thought to affect more than 50,000 people in the United States and Europe alone2. The underlying genetic cause is a trinucleotide CAG expansion in the huntingtin gene (HTT), discovered as a genetic marker by James Gusella from Massachusetts General Hospital in 1983, which results in the production of a mutant huntingtin protein (mHTT) with a toxic poly-glutamine (polyQ) tract3,4. However, despite decades of research and clinical trials, no successful therapy has yet been developed. The “typical” onset of HD is between the ages of 40–50 years, but up to 15% of cases have very late onset and don’t show clinical symptoms until after the age of 60 years5. In a recent meta-analysis of studies investigating cases of late-onset HD (LoHD, defined as onset after 60 years of age), more than 90% of patients had CAG repeat lengths of ≤446. One of the more interesting observations in HD is that while there is a well-known correlation between the length of the polyQ repeat tract and the onset and severity of the disease, there is substantial variability within individual patients. For example, in patients with mid-range repeat lengths (defined here as between 40 and 50), disease onset can vary by 60 years in any individual patient5. This means that many patients who are carriers of polyQ tracts that predispose to the development of the disease can live for decades in a “presymptomatic” state7. What controls the onset of clinical symptoms remains currently unknown, and complicates the prognostic evaluation of HD patients.
Although historically considered a monogenic disease, extensive research into the underlying pathology of HD suggests that the mechanisms leading to disease onset and progression are more complex than originally thought. Many different technologies have been used to look at the molecular changes underlying disease progression in HD, including gene expression, proteomics, metabolomics, network analysis, genomics and single nucleotide polymorphism (SNP) profiling8–13. Recently, epigenetic approaches have emerged as a promising new tool for assessing pathology-related changes14. Most epigenetic studies in HD have focused on looking at genome-wide histone modifications (acetylation, methylation) or histone modifications at specific loci related to HD15–18. While these approaches have provided interesting insight into the disease, they have yielded often conflicting results and shown inconsistencies between mouse models and human disease14,19,20. Indeed, due to the global nature of these types of epigenetic analyses, they may lack the sensitivity to discriminate the subtler changes associated with disease onset and progression. As such, a consensus picture of epigenetic deregulation in HD using histone modification readouts has yet to materialize. However, not all molecular mechanisms associated with epigenetic regulation have been assessed in the context of HD. An important aspect of epigenetic regulation is at the level of 3-dimensional (3D) genomic architecture21.
The 3D organization of the genome reflects the heterogeneous effects of external environmental cues and inputs, and can be empirically measured by the assessment of chromosome conformations or when several conformations are measured concomitantly, a chromosome conformation signature (CCS)21. CCSs can be thought of as the molecular barcode that gives a readout of the epigenetic landscape of a given cellular population22,23 To date, the evaluation of CCSs in HD has remained unexplored. Given the central role of mHTT in the development of HD, we hypothesized that regulatory differences in genomic architecture at the HTT locus may exist between diseased individuals and healthy, unaffected controls.
We used EpiSwitch, an established proprietary industrial platform for monitoring CCSs, to assess chromatin architecture differences between pre-symptomatic and symptomatic HD patients and healthy, unaffected individuals. EpiSwitch readouts provide high resolution, reliable and high throughput detection of CCSs while simultaneously meeting the high bar of industry standards for quality control21. As such, this technology represents a powerful tool for screening, evaluation and monitoring of CCS in human disease24. This platform has been successfully utilized as a biomarker modality to stratify patients in the context of a variety of other diseases25–30, including as a non-invasive blood based biomarker for neurodegenerative conditions31,32.
All blood samples were obtained from National BioService, LLC, a research biobank operating in compliance with the requirements of the International Society for Biological and Environmental Repositories. In total, 20 blood samples were used in this study; 10 healthy control (HC) samples (CAG repeats, n<35), and 10 HD samples (CAG repeats, n>39). For the HD samples, 7 were from symptomatic patients (HD-Sym) and 3 were from presymptomatic patients who had a diagnosis of HD but did not yet show any clinical symptoms (HD-Pre). One HD patient was taking tetrabenazine and one patient was taking sertraline. All samples were negative for human immunodeficiency virus, hepatitis B virus, hepatitis C virus and syphilis (Supplemental Table 1).
We wanted to identify chromosome conformations that differed between healthy controls (low CAG), presymptomatic HD patients (high CAG, no disease manifestation) and symptomatic HD patients (high CAG, disease manifestation). We focused on a ~225 kb region surrounding the HTT locus from (chr4: 3,033,588 to 3,258,170 as annotated in hg38) for our analysis. Using the CAG repeat expansion tract in exon 1 of HTT (chr4: 3,054,162 to 3,095,930) as the anchor point (“Anchor”), we defined five genomic zones surrounding the anchor to look at chromosome conformations that varied between sample groups (Figure 1 and Supplemental Table 2). These Zones were chosen based on: the presence of potential EpiSwitch anchoring sites, the presence of known disease-related SNPs (HD and other diseases), and the enrichment of known histone modification sites (H3K4me3, H3K36me3, and H3K27ac) in HD as found in the GWASdvV2 database (http://jjwanglab.org/gwasdb) (Figure 1).
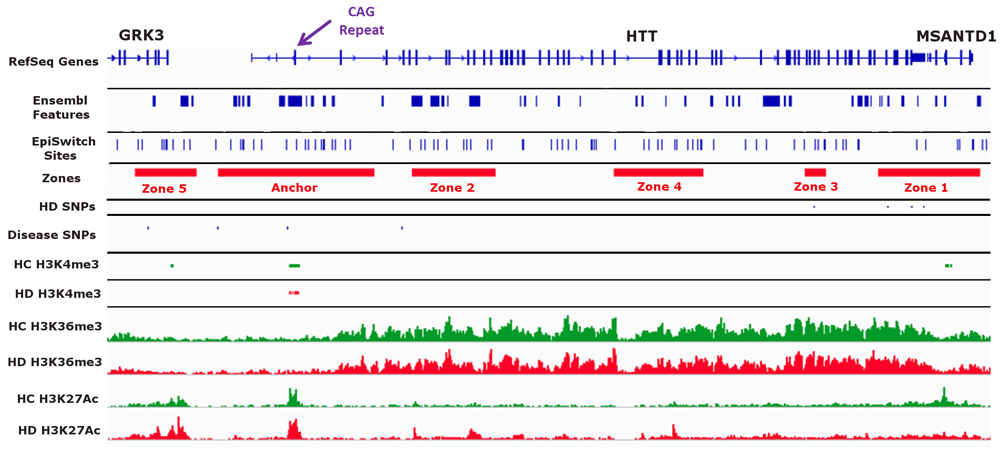
A visual overview of the genomic region investigated in this study. A ~225 kb region on chromosome 4 spanning the HTT locus was investigated. The anchor point (“Anchor” in track 4) was defined as a ~42 kb region spanning the CAG repeat tract in exon 1 of HTT (purple arrow at the top of the figure). We defined five Zones (Zones 1 -5 in track 4) based on overlap with EpiSwitch sites (track 3), SNPs related to Huntington’s disease (HD) (track 5) or other diseases (track 6), and observed methylation and acetylation (H3K4me3, H3K36me3 and H3K27Ac) differences between healthy control (HC) and HD (tracks 7 through 12).
A search of the NCBI Gene Expression Omnibus database for previously reported HD epigenetic data was performed in February 201833. Peak-called ChIP-seq data for H3K4me3 from 12 (6 HD and 6 control samples) post-mortem prefontal cortex brain samples (bed format) was obtained (GSE68952)34. In addition, Bigwig tracks of ChIP-seq data for H3K27ac and H3K36me3 from HD iPSC-derived neural cell lines and control cell lines were also downloaded (GSE95342)35. The data tracks were loaded into the Integrative Genome Viewer (IGV)36 version 2.4 alongside the EpiSwitch and reference sequence annotations. Both visual and programmatic (BEDtools) comparisons were performed on the HTT locus to identify the five zones of interest.
Oxford BioDynamics proprietary EpiSwitch pattern recognition algorithm was used to identify high probability chromatin folding interactions with one “end” occurring in the anchor zone proximal to the CAG repeats and the other in any of the five zones of interest. A total of 61 interactions matched these criteria, and for practical reasons, 20 interactions were selected to cover interactions between the anchor site and all the zones of interest. Oxford BioDynamics automated primer design application was used to design oligonucleotide pairs that amplified the expected DNA sequence caused by the interaction when subjected to the chromosome conformation capture (3C) assay.
3C and detection by PCR were performed as described previously25,28,30,37. Chromatin with intact chromosome conformations from 50 µl of blood sample from each patient sample was extracted using the EpiSwitch assay following the manufacturer's instructions (Oxford BioDynamics Plc). Quality control on all samples was done using the detection of a chromatin loop at the MMP1 locus, a historical internal control for 3C analysis30. Pooled 3C libraries for each of the sample types were generated to provide a generalized population sample for each of the C sample subgroups. Real-time PCR experiments were performed in accordance with the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines38. Real-time PCR was performed with SYBR green with the CFX-96 (Bio-Rad) machine to identify the interactions with differing PCR product detection patterns between the sample types39. Oligonucleotides were tested on control templates to confirm that each primer set was working correctly. A full list of the primers and PCR conditions used in this study can be found in Supplementary File 1. The final nested PCR was performed on each sample in triplicates for the follow up data on individual patients with HD. This procedure permitted the detection of limited copy-number templates with higher accuracy. All PCR-amplified products were monitored on the LabChip® GX from Perkin Elmer, using the LabChip DNA 1K Version 2 kit (Perkin Elmer) and internal DNA markers were loaded on the DNA chip according to the manufacturer’s protocol using fluorescent dyes. Fluorescence was detected by laser and electropherogram read-outs translated into a simulated band on gel picture using the instrument software. The threshold of detection for the instrument was set by the manufacturer from 30 fluorescence units and above. All raw gel images for the PCR assays done in this study as well as a description of each set of comparisons can be found in Dataset 140.
Data analysis was performed in R (language and environment for statistical computing) version 3.5.1 (https://www.r-project.org)41. This included stats (version 3.6.0) and dplyr (version 0.7.6) packages for t-tests and R2 analysis & a ggplot2 (version 3.0)42 package for boxplots and regression plots.
HC and HD samples were age (average 36.9 years for HC and 35.3 years for HD) and sex matched (10 male and 10 female), with the majority (70%) of HD cases being symptomatic (Table 1). All samples were from non-Hispanic or Latino whites. Average CAG repeats lengths were 25.7 for HC and 44.2 for HD (Table 1, Figure 2). There was no statistical difference in CAG repeat length between HD-Pre and HD-Sym (Figure 2). The average age at diagnosis for HD samples was 35.3 years and the average disease duration was 3.8 years with 7 out of 10 patients reporting symptoms of irritability, chorea, or both (Table 1).
| Variable | Healthy controls (N=10) | Huntington's disease (N=10) |
|---|---|---|
| Gender | ||
| Male (N, (%)) | 5 (50) | 5 (50) |
| Female (N, (%)) | 5 (50) | 5 (50) |
| Ethnicity | ||
| Non-Hispanic or Latino | 10 (100) | 10 (100) |
| Race | ||
| White (N, (%)) | 10 (100) | 10 (100) |
| Huntington's Type | ||
| Symptomatic (N, (%)) | N/A | 7 (70) |
| Asymptomatic (N, (%)) | N/A | 3 (30) |
| CAG repeat length (average, (SD)) | 25.7 (5.4) | 44.2 (2.6) |
| Age at diagnosis (average, (SD)) | N/A | 34.9* (8.1) |
| Age at sample collection (average, (SD)) | 36.9 (12.8) | 35.3 (10.0) |
| Disease duration (years) (average, (SD)) | N/A | 3.8 (1.9)** |
| % Reporting irritability | N/A | 70 |
| % Reporting chorea | N/A | 50 |
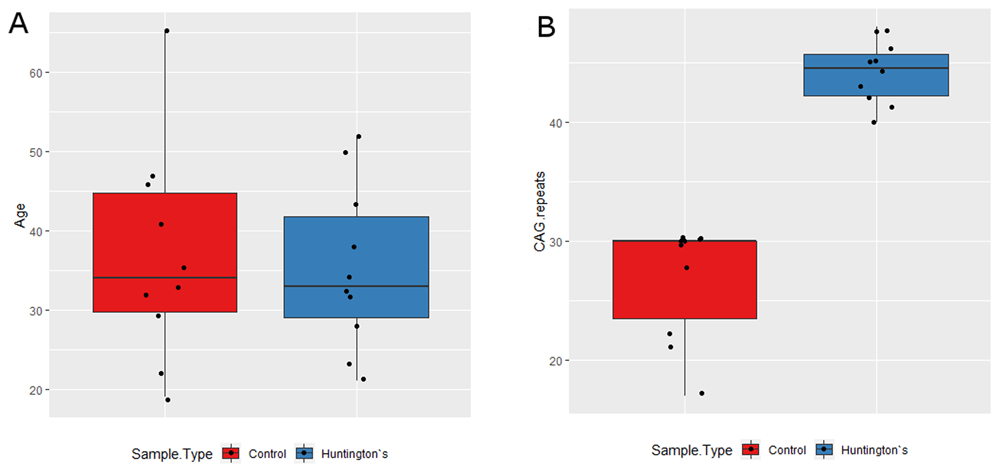
(A) The HC (red) and HD (blue) samples showed no statistical difference in age. HD-Pre patients were younger (average age = 25.3) than HD-Sym patients (average age = 39.6) (p = 0.02). There was a moderate negative relationship between disease duration and CAG repeat size and a moderate positive relationship between age at diagnosis and CAG repeat size, though neither were statistically significant (data not shown). (B) There was a statistically significant increase in CAG repeat length in HD patients (blue) relative to HC (red) (p = 1.08 E-7). There was no statistical difference in CAG repeat length between HD-Pre and HD-Sym (p = 0.09) (data not shown).
Of the 20 interactions that were evaluated (Supplemental Table 3), we identified nine informative interactions. We identified two constitutive interactions and seven conditional interactions which were present in HD, but not healthy controls. Of the seven conditional interactions, three were present only in HD-Sym, and absent in HD-Pre.
All samples passed internal QC analysis for the MMP1 interaction (Supplementary Figure 1 and Supplementary Figure 2). Two constitutive (identified in all samples) chromatin loops were identified. Both loops were between the Anchor and Zone 2 with the first loop spanning 28 kb and the second loop spanning 34 kb (Figure 3).
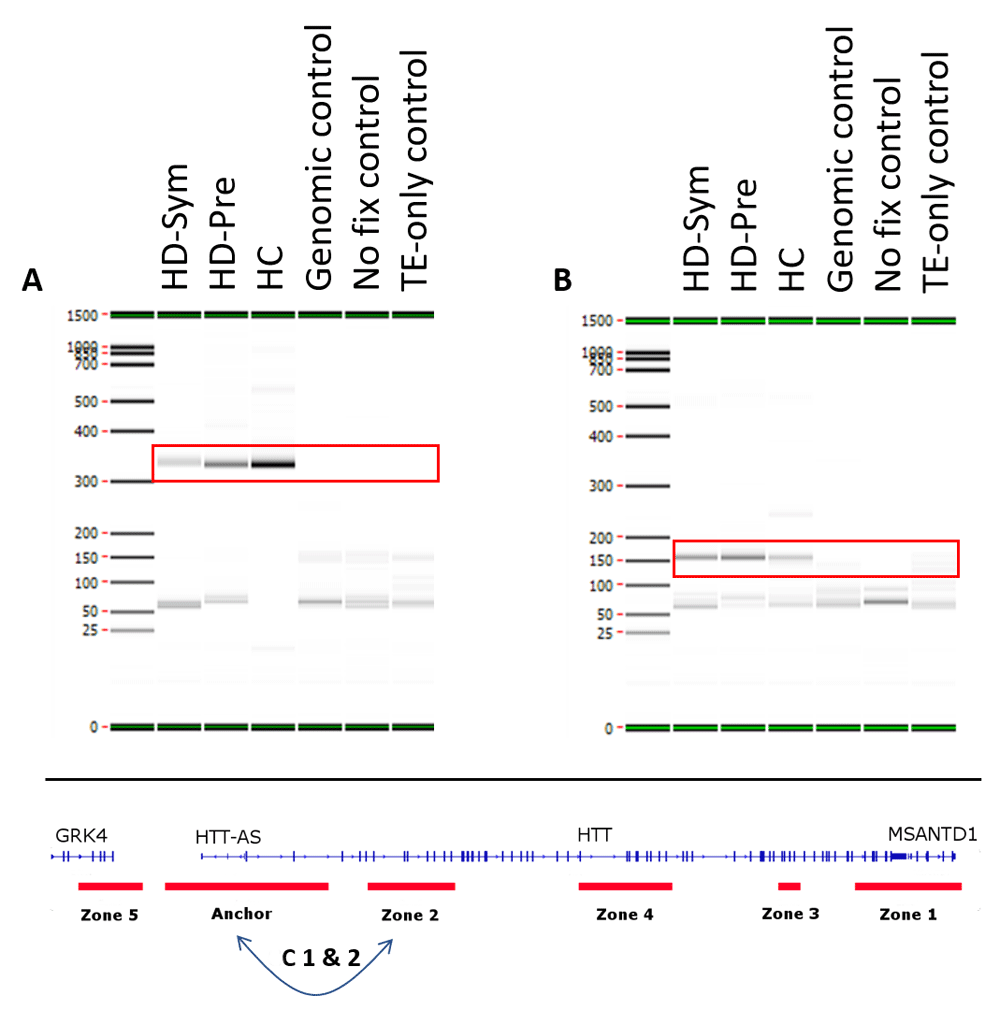
Two constitutive interactions occurring in all patients (symptomatic Huntington’s disease (HD-Sym), Pre-symptomatic HD (HD-Pre) and healthy control (HC)) (red boxes) were observed in this study. Both interactions (C1 & C2) were between the anchor and zone 2 with C1 (A) spanning 28 kb and C2 (B) spanning 34 kb. TE=Tris-EDTA.
We identified seven conditional chromosome conformations that could discriminate between the different patient subgroups evaluated in this study. Specifically, we identified two chromosome interactions that were present in HC, but absent in all HD samples (Figure 4). The first interaction (I1) spanned the anchor and zone 4 and covered 77 kb while the second interaction (I2) spanned the anchor and zone 3 and covered 140 kb. We also identified two chromosome interactions that were present in HC and HD-Pre, but absent in HD-Sym samples. Both interactions (I3 and I4) spanned the anchor and zone 4, and covered 92 kb and 104 kb, respectively (Figure 5). Last, we identified three chromosome interactions that were present in HD-Sym samples, but absent in HD-Pre and HC samples. The first of these three conditional interactions (I5) spanned the anchor and zone 3, covering 122kb. Notably, this interaction included a SNP (rs362331) known to be a factor in the predisposition to develop HD. The second and third conditional interactions (I6 and I7) spanned the Anchor and Zone 1 and covered 185 kb and 174 kb, respectively (Figure 6). Last, we tested the absence or presence of all conditional interactions in individual HD samples. In the HD-Sym samples, we found the presence of at least one of the conditional markers (I5, I6 and I7) in six out of seven samples (Figure 7 and Supplementary Figure 3–Supplementary Figure 5). The odds ratios of each interaction being associated with symptomatic HD presentation were 30, 9 and 16 for I5, I6 and I7, respectively. A summary of all the interactions that were evaluated in this study are shown in Figure 8 and Supplementary Figure 6.
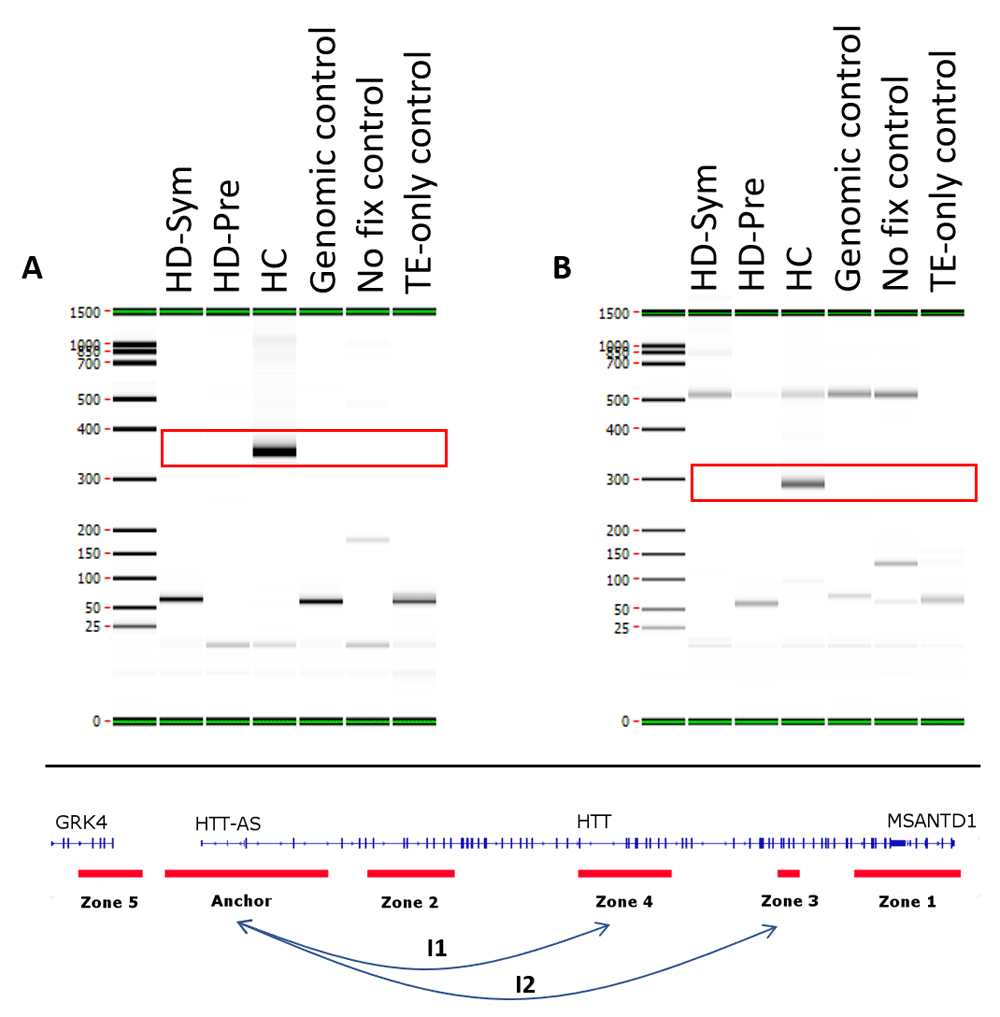
Two conditional interactions (red boxes) were observed in HC patients only and were absent from all Huntington’s disease (HD) patients. (A) The first interaction (I1) occurred between the Anchor and Zone 4 and spanned 77 kb. (B) The second interaction (I2) occurred between the Anchor and Zone 3 and spanned 140 kb. TE, Tris-EDTA.
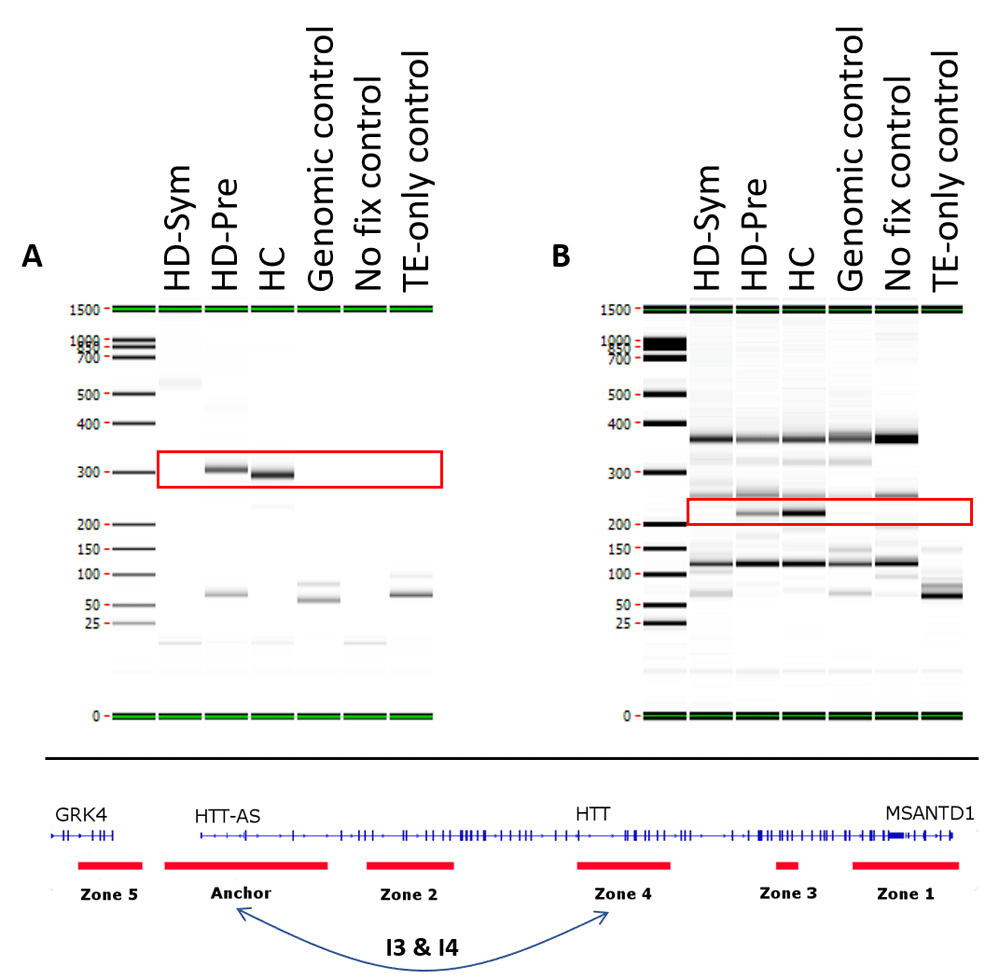
Two conditional interactions (red boxes) were observed in healthy control (HC) patients and pre-symptomatic HD (HD-Pre) patients, but were absent from symptomatic HD (HD-Sym) patients. Both interactions (I3 & I4) occurred between the anchor and zone 4 and spanned 92 kb (A) and 104 kb (B), respectively. TE, Tris-EDTA.
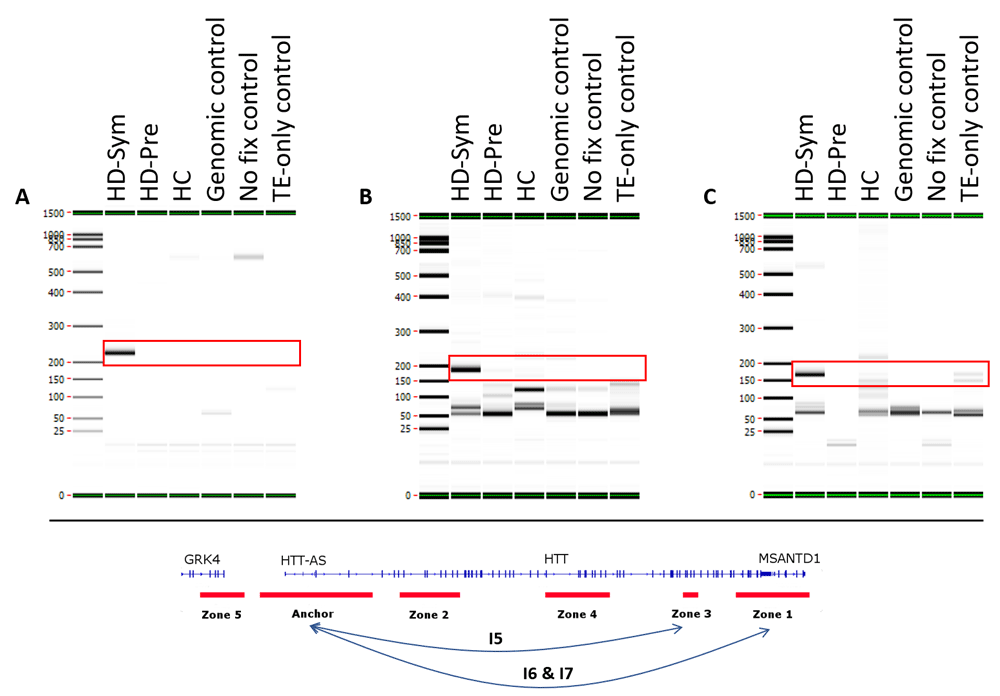
Three conditional interactions (red boxes) were observed in symptomatic HD (HD-Sym) patients only and were absent from HC and pre-symptomatic (HD-Pre) patients. (A) The first interaction (I5) occurred between the anchor and zone 3 and spanned 122 kb, including a HD-associated SNP known to be involved in disease progression. (B and C) The second and third interactions (I6 & I7) occurred between the anchor and zone 1, and spanned 185 kb and 174 kb, respectively. TE, Tris-EDTA.

In six out of seven individual HD-Sym samples, the presence of at least one of the three conditional interactions (I5, I6 and I7) was observed. I5, the interaction spanning the region that contains the rs362331 SNP, was observed in the greatest number of samples (5/7).
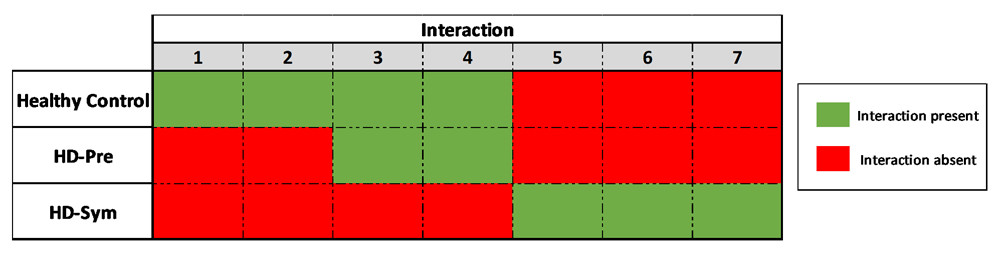
Overview of the chromosomal conformation changes associated with the progression of HD. As patients progress from presymptomatic stages to symptomatic diseases, discrete, measurable and discriminating changes in the genomic architecture at the HTT locus are observed.
While it is well-known that individuals with greater than 39 CAG-repeats will get HD, the clinical onset of disease varies widely amongst individual patients and the factors that influence when the disease manifests clinically are less well characterized. Here we used EpiSwitch, an industrial platform for assessing chromatin architecture, to evaluate the epigenomic landscape of the HTT locus in HD patients and healthy, unaffected controls. We identified a set of seven interactions that when taken together as a CCS, could differentiate HD from unaffected controls and more importantly, could differentiate between presymptomatic and symptomatic HD patients. One of these interactions, specific for symptomatic HD, contains a SNP (rs362331) shown to be associated with a predisposing disease haplogroup. When taken together, these results provide an initial indication that a simple, non-invasive blood-based test evaluating a CCS can serve as a surrogate biomarker for assessing disease progression in HD.
While it is known that the poly-Q repeat tract expansion and production of mHTT are the underlying causes of HD, the molecular events leading to the development of clinical symptoms are less well characterized. Several studies have looked at SNPs within the HTT locus as a potential contributor to disease onset. One recent SNP genotyping study of HD patients identified ~41 SNPs heterozygous in at least 30% of the patients, including the rs362331 C/T SNP in exon 50 of the HTT gene43. Perhaps more biologically relevant is that when the rs362331 SNP is allele-selectively knocked down using anti-sense oligonucleotides, siRNAs or miRNA, a dramatic reduction in the levels of mHTT protein is achieved both in vitro and in vivo, suggesting that this SNP and its surrounding genomic landscape play an important role in regulating mHTT levels44–46. In this study, we observed a chromosome conformation (I5) that was present in HD-Sym patients, was absent in HD-Asy and HCs, and overlapped with the rs362331 SNP. While requiring further study, this observation raises the interesting possibility that the production of neurotoxic mHTT in patients that have increased poly-Q tracts and a genetic predisposition to the early development of HD by the presence of the rs362331 SNP may be regulated at the level of higher-order chromatin structure. Another outstanding question in HD is how the disease is inherited in cases where neither parent has received a diagnosis. The two main prevailing hypotheses posit that 1) the carrier parent could have passed away from another factor before the onset of the disease and 2) “unstable” CAG repeat tracts expand with each generation. A third possibility also exists, in that at mid-range (35-50) repeats, individuals could be carriers without manifestation of the disease, but their progeny might be unable to compensate for the genetic defect through undefined mechanisms and will develop the disease. The HD patients evaluated in this study all had CAG repeats in this mid-range, raising the possibility that potential compensatory mechanisms in disease development may be mediated through differences in genomic architecture.
HD is rare, in that there exists a simple test to definitively diagnose the disease, HTT gene sequencing and measurement of CAG repeat number. For clinical care and clinical trials, there are also several tests to measure disease severity, such as the Unified Huntington’s Disease Rating Scale, the Shoulson–Fahn Scale, and the Mini–Mental State Examination47–49. While these assessments measure different elements of an HD patients physical and mental well-being as a surrogate for disease severity, they are all subjective in nature and most are not specific for HD. What is missing are concrete molecular tools to monitor disease progression.
As of the time of this writing, there are 22 therapeutic agents for treating HD in different stages of preclinical and clinical development, half of which are in Phase 2 or Phase 3. Once further validated, the CCS reported here could be used in clinical trials as a surrogate outcome biomarker to assess the therapeutic efficacy of the drug in question. In addition to monitoring a symptomatic patient’s response to a particular therapy in clinical trials, another advantage of the approach described here lies in the information that can be obtained for pre-symptomatic patients. For most patients with HD, the pre-symptomatic period can last decades. Five of the seven (i3–i7) interactions identified here clearly separate presymptomatic HD patients from symptomatic ones, and when further validated could serve as an “early warning” indicator test for the onset of HD symptoms in presymptomatic carriers.
This study gives first evidence of detectable conditional differences in chromatin architecture specific for the manifestation of HD and correlated with known disease haplotypes. The major strength of this study lies in its unique approach, which is based on the latest developments in understanding the regulatory role of genomic architecture. While there have been several historical studies in HD aimed at developing disease progression biomarkers based on clinical, imaging and molecular measures50, to the best of our knowledge this is the first time that the assessment of higher-order chromatin structures in a clinically accessible biofluid has been applied in HD. With the successful application of EpiSwitch in another neurodegenerative condition, amyotrophic lateral sclerosis, as well as other non-neurological conditions such as melanoma, diffuse large B-cell lymphoma, chronic myelogenous leukemia, breast cancer, and rheumatoid arthritis, the results presented here further validate the use of regulatory conditional CCS as disease-related biomarkers25,28–31. A notable limitation of this study was the small sample size.
While the data presented here offer a novel insight into the clinical progression of HD, this study was intended to be a proof-of-concept and not powered for statistical significance. A follow-up study using a larger patient cohort will be required for validation of these initial results.
Dataset 1. Raw gel images for all PCR reactions performed in this study. Also included is a guide to the contents of the images. DOI: https://doi.org/10.5256/f1000research.15828.d22287340.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Reviewer Comment to response #1: If this is the case than this should clearly be reflected in the title. The title is now misleading. I am not stating that the methods are invalid, or not properly done, but I remain of the opinion that the sample size is too small, and the results are too variable to draw any conclusion. See Figure 3 and my previous comment #9.
Author Response to Reviewer Comment to response #1:
We agree and have updated the title to reflect the nature of the study and the extent to which we can draw conclusions from it.
Reviewer Comment to response #2: I still think the text should be changed to something like: “HD is a monogenic disease but there are many other genetic, environmental and unknown factors that can influence onset and progression of the disease”.
Author Response to Reviewer Comment to response #2:
We agree and have updated the text to reflect this.
Reviewer Comment to response #4: Carini et al used 59 patients, Salter et al used 74 unblinded patient samples, Yan et al used 58 patients. Proof of concept of this analysis was done in these papers. However, if this analysis is useful as a tool to assess disease progression cannot be deducted from the data presented in this paper.
Author Response to Reviewer Comment to response #4:
We agree and have updated the text to reflect this. Specifically, we have modified the language in the Abstract-Conclusions, Discussion-Results summary and Discussion-Strengths and limitations to directly address the reviewers concern.
Reviewer Comment to response #1: If this is the case than this should clearly be reflected in the title. The title is now misleading. I am not stating that the methods are invalid, or not properly done, but I remain of the opinion that the sample size is too small, and the results are too variable to draw any conclusion. See Figure 3 and my previous comment #9.
Author Response to Reviewer Comment to response #1:
We agree and have updated the title to reflect the nature of the study and the extent to which we can draw conclusions from it.
Reviewer Comment to response #2: I still think the text should be changed to something like: “HD is a monogenic disease but there are many other genetic, environmental and unknown factors that can influence onset and progression of the disease”.
Author Response to Reviewer Comment to response #2:
We agree and have updated the text to reflect this.
Reviewer Comment to response #4: Carini et al used 59 patients, Salter et al used 74 unblinded patient samples, Yan et al used 58 patients. Proof of concept of this analysis was done in these papers. However, if this analysis is useful as a tool to assess disease progression cannot be deducted from the data presented in this paper.
Author Response to Reviewer Comment to response #4:
We agree and have updated the text to reflect this. Specifically, we have modified the language in the Abstract-Conclusions, Discussion-Results summary and Discussion-Strengths and limitations to directly address the reviewers concern.