Keywords
hyaluronic acid, chondroitin sulfate, protein-polymer assembly
This article is included in the Nanoscience & Nanotechnology gateway.
hyaluronic acid, chondroitin sulfate, protein-polymer assembly
The introduction now clarifies the differences between covalently tethering functional moieties that facilitate supramolecular network formation, and supramolecular networks of unfunctionalized hyaluronic acid (reviewer 1). The utility and reasons to use BSA were also clarified (reviewer 2).
The introduction and the results/discussion sections were modified to clarify the nomenclature of non-covalent interactions of HA and CS, and distinguish between the importance of these interactions for each polymer in physiology. Figure 1 was updated (reviewer 1 and 2). ESI figures were moved to the main manuscript from the Extended data. We include the point made by reviewer 2 on CS/BSA interactions, as well as underscoring the point that denatured protein was key to forming nanostructures or gels.
Protein concentration was varied and the rheological properties were measured by new authors June Y. Park and Vijay K. Rana to show tunability. SEM images of the dried hydrogels were added. Further text was included to contextualize the qualitative nature of Figure 3D. Electrostatically-driven crosslinking of the polymer-protein systems interfered with the voltage signal of the cantilever during AFM measurements (reviewer 1). Addition of solvents changed gel properties via diffusion of polymer/protein into solvent or diffusion of water out of the system. The Figure 6 caption was edited to clarify that rhodamine under visible light is pink (reviewer 1).
The first reported methods describing the formation of HA/BSA gels resulted in gels that were sensitive to the force exerted during mixing. These methods have been expanded to be more reproducible from person-to-person. The title was modified to address reviewer comments and better represent the scope of the work. Minor mistakes in the reported concentrations as well as instrument specifications were corrected. Minor changes in the text in the abstract were made to address comments by both reviewers.
Funding from additional author JYP was added.
To read any peer review reports and author responses for this article, follow the "read" links in the Open Peer Review table.
A major paradigm that has dominated the drug delivery and tissue engineering communities is the development of bio-inspired hydrogels that mimic the intermolecular interactions and mechanical properties of physiological tissue. Glycosaminoglycans (GAGs) are polysaccharides that are critical structural components of the brain extracellular matrix (ECM). One particularly abundant brain GAG, hyaluronic acid (HA) (Figure 1A), has been made into many covalently modified derivatives that have been widely explored in drug delivery and tissue engineering1,2. Noteably, HA is the only purely supramolecular brain GAG3–7, i.e. weaker and reversible non-covalent interactions dominate over covalent bond formation. Many groups have reported HA-based materials that consist of chemically functionalizing the polymer backbone with moieties that can facilitate formation of supramolecular (such as in physiology) or covalent networks8,9. Networks formed from unfunctionalized HA, however, have received considerably less attention in the literature. Furthermore, other ECM components, including the more abundant protein-linked GAG chondroitin sulfate (CS) (Figure 1A), have been comparatively under studied for biomaterial applications10.
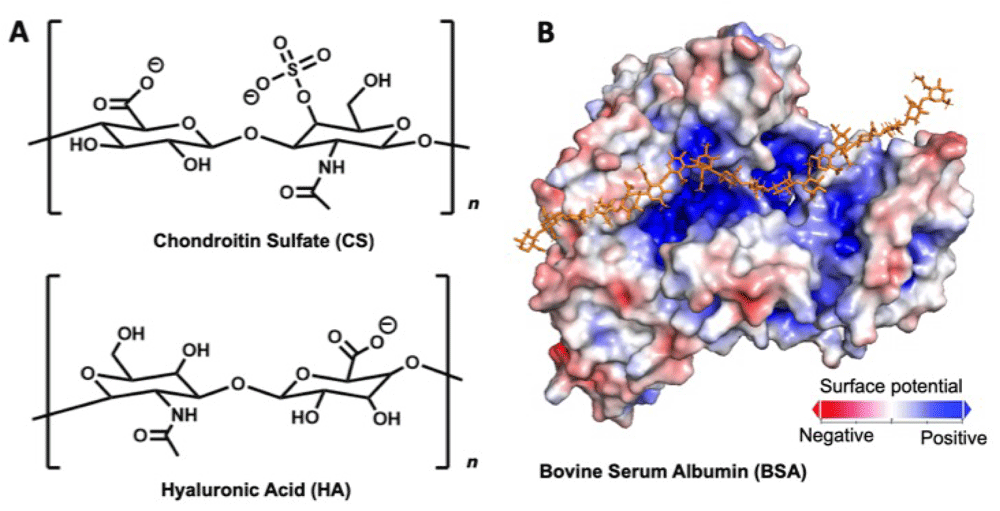
(A) Structures of chondroitin sulfate (CS) and hyaluronic acid (HA). (B) Relative surface charge densities of bovine serum albumin (BSA). BSA’s binding pocket is indicated with the example HA chain.
In this work we examine the intermolecular interactions of two major biopolymer components of the brain, HA and CS, with the model protein bovine serum albumin (BSA) (Figure 1B). While BSA is not normally found in the brain and its physiological relevance is limited, we demonstrate herein the potential for misfolded or unfolded protein to form supramolecular complex with ECM polymers to drastically change their mechanical properties. Heilshorn and colleagues previously reported on supramolecular hydrogels formed via protein interactions with polysaccharide backbones11–13. In this work we exploit interactions between BSA and GAG polymers and induce changes in protein folding to generate structurally diverse protein-glycosaminoglycans complexes. We examine how these negatively charged biopolymers interact and self-assemble with BSA, and develop non-covalent systems formed from competing HA/BSA, HA/CS, and CS/BSA interactions.
All starting materials were purchased from Sigma Aldrich and used as received unless stated otherwise.
The crystal structure of bovine serum albumin (BSA) was downloaded from Protein Data Bank (ID:3V03). The second chain of the homodimer in the crystal structure was removed to display BSA in monomeric form. Using the Adaptive Poisson-Boltzmann Solver (APBS) tool available through PyMOL v2.2.2, the electrostatic surface potential was calculated under default parameters14. The section of the protein surface showing the previously-described GAG binding pocket was rendered with PyMOL.
A 10 wt% chondroitin sulfate (C9819 Sigma) solution was prepared by mixing the polymer powder in Milli-Q H2O (18 mΩ) at room temperature overnight. Bovine serum albumin (BSA; 50 mg/mL) was added to the solution and rigorously mixed for 12 h (1000 RPM) at room temperature. Lower total concentrations of polymer and protein took more than 12 h to form particles.
4 wt% hyaluronic acid (1.5–1.8 MDa; 53747 Sigma) solutions were prepared by mixing the polymer powder in Milli-Q H2O (18 mΩ) at 40°C for 40 h. The solution was sealed and stored at 4°C until further use. To electrostatically crosslink HA, BSA was added and partially denatured via mechanical force or heat. Various concentrations of protein were added to the viscous polysaccharide solution and mixed vigorously with a metal spatula for several minutes. The samples were sealed until they appeared transparent, typically for at least 24 h. BSA denaturing could also be accomplished with heat by incubating samples at 50°C until the solution turned opaque (typically 2 h). It was observed that BSA retaining its folded structure would not result in gel formation (see results and discussion). To form HA/CS/BSA systems, dry chondroitin sulfate (10 wt%) was added along with the dry BSA into HA solution and the same procedure as HA/BSA was used. Poly(caprolactone) (PCL) blends were formed by mixing poly(caprolactone) diol (Mn = 2 kDa; Sigma 189421; 5 wt%) with poly(caprolactone) diol melt (Mn = 550 Da; Sigma 189405) and mixing at 700 RPM at 50°C overnight. A specified amount of rhodamine B was added to the blend for ultraviolet–visible spectroscopy (UV-Vis).
All rheological sweeps were conducted on an Discovery HR-2 Rheometer (TA Instruments, New Castle, DE, USA) with a 40 mm parallel plate geometry at 20°C Zero gap, rotational mapping (precision bearing mapping; 2 iterations), geometrical inertia, and friction calibrations were done prior to each use of the rheometer. Samples were loaded onto the rheometer with a 600–1000 µm loading gap. A water trap was placed to prevent dehydration. Amplitude sweep were conducted to determine a strain in the linear viscoelastic region.
Dynamic light scattering (DLS) measurements were carried out on a Malvern Zetasizer NS90 instrument at room temperature and standard settings. Samples were analysed in a 1.5 mL PS cuvette (Fisher Brand).
Transmission electron microscopy (TEM) was carried out on a FEI Philips Tecnai 20. Samples were prepared on holey carbon grids by pipetting 1 µL of desired aqueous solution and allowing it to evaporate under ambient conditions (drop-casting). Particle size distributions were calculated by counting the diameters of more than 100 particles.
UV-Vis spectroscopy was performed using a Mikropack DH-2000 UV-Vis-NIR Halogen light source and an OceanOptics USB2000 Fiber Optic Spectrometer. Spectra from 375 nm to 750 nm were recorded at 150 ms integration time and time intervals of 60 s.
Scanning electron microscopy (SEM) was carried out on a Tescan MIRA3 SEM. Samples were freeze dried in small volumes and thin flakes were carefully mounted onto sample stubs covered in carbon conductive adhesive tapes. Each sample was then placed into a platinum sputtering system and sputter-coated to 10 nm thickness. To load the sample, the SEM chamber was vented and the sample stubs were loaded and tightened into places. After closing the sample compartment and allowing the system to reach vacuum, images were captured using operating voltage ranging from 10–30 kV.
To estimate brain chondroitin sulfate (CS) levels, all articles cited as using the Blyscan assay to measure sulfated GAGs were searched against the keywords “brain” and “neural”. A total of 7 articles were found meeting the criteria of measuring sulfated GAGs in mammalian brain tissue. A separate literature search of hyaluronic acid (HA) measurements identified 2 articles. Reported concentration values were converted to molarities, representing the moles of disaccharide repeat units per volume of native brain tissue, assuming a brain density of 1.04 g/ml15. In cases where brain weight was reported as dry mass instead of native tissue, masses were converted by assuming that 77% of brain weight is water16.
In this work we explored the self-assembly of charged proteins with negatively-charged polysaccharides endogenous to the human brain. To facilitate supramolecular crosslinking of these polysaccharides, we introduced BSA, which has electrostatic binding pockets complementary to anionic GAGs, analogous to the binding interfaces of ECM proteins.
BSA and CS polymer solutions were mixed rigorously overnight and allowed to self-assemble into nanostructures. Two distinct populations of particles were observed to form (Figure 2B-D, Dataset 117), and were dissimilar to the flocculation of BSA aggregates alone. The smaller particles were characterized with dynamic light scattering (DLS) and transmission electron microscopy (TEM). DLS yielded an average particle diameter (D) of 51 ± 3 nm. These structures were stable for at least 2 days in the parent suspension (Figure 2E). DLS autocorrelation data suggested that a large diameter species may also be present in the solution (Figure 2F). TEM was used to characterize these self-assembled microparticles (Figure 2D). The analysis of these micrographs indicated the presence of two distinct populations of assembles, with the mean diameter of D = 60 ± 10 nm, that is consistent with DLS experiments, and an additional one of D = 1.5 ± 0.5 µm. In the brain, CS is covalently scaffolded onto protein cores (e.g. aggrecan18) and binds with many ECM proteins through non-covalent, supramolecular interactions19. The non-covalent interactions between CS and BSA described here, potentially owed to electrostatic interactions or phase separation, could provide insight into driving forces that contributes to GAG aggregation and nanostructure formation in vivo.
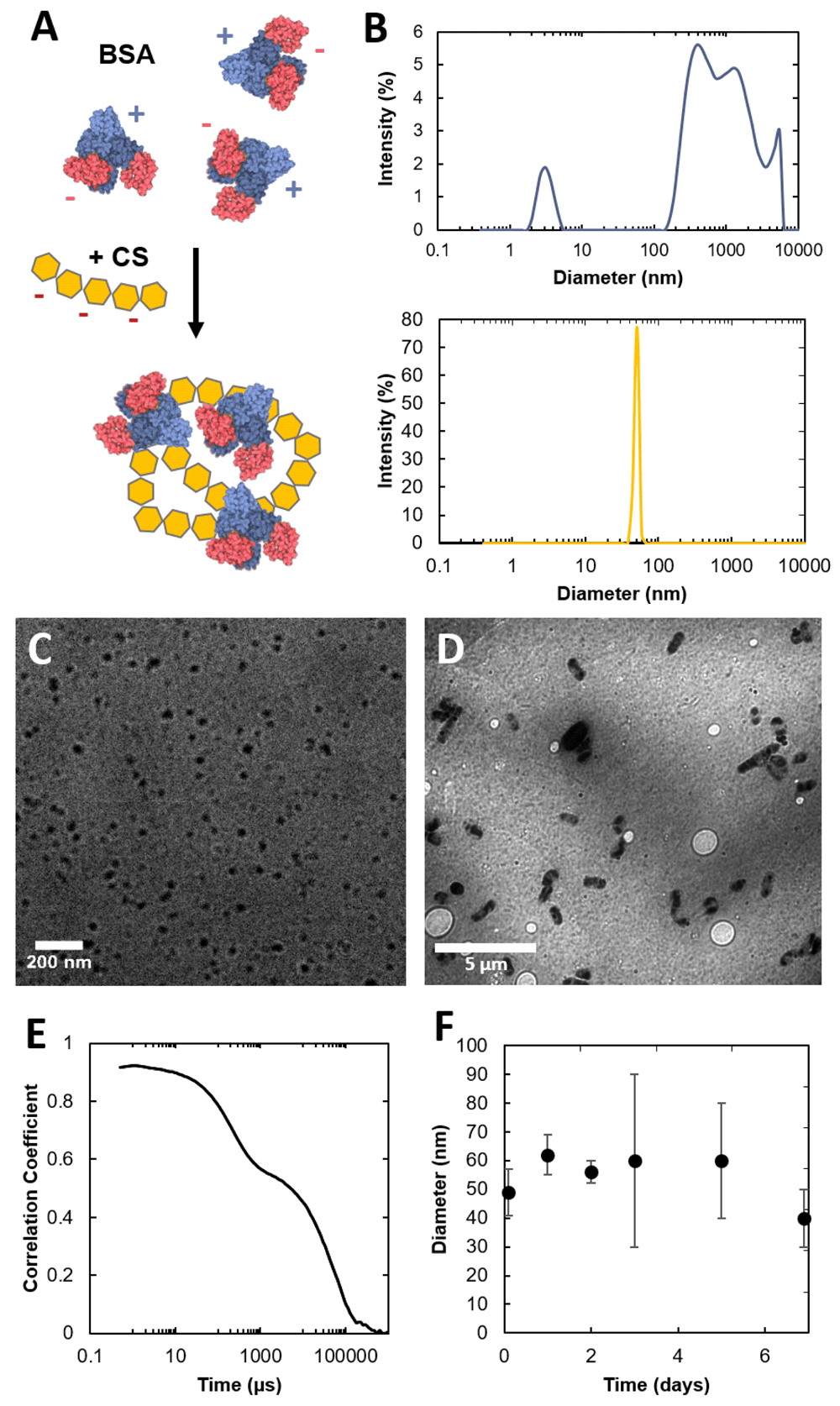
(A) Schematic of the formation of dense CS-BSA particles. (B) DLS size plot of dynamic BSA aggregates (top) and CS-BSA particles (bottom). (C–D) TEM image of CS-BSA nanoparticles (C) and microparticles (D). (E) Autocorrelation function of CS/BSA NPs. (F) Time resolved DLS of CS/BSA NPs. Dataset 1: Dynamic light scattering and transmission electron microscopy data17.
We then turned our attention to HA, a linear high molecular weight polysaccharide that is the only supramolecular GAG in human physiology. HA and BSA were mixed and the electrostatic interactions between the charged moieties of the GAG and the unfolded BSA protein led the solution to self-assemble into a supramolecular gel (Figure 3, Dataset 117). Solutions of HA alone were highly viscous but did not show gel-like properties. Oscillatory rheological measurements were used to probe the mechanical properties of this supramolecular gel. After introduction of partially denatured BSA, electrostatic-driven network percolation resulted in a major stiffening effect and the formation of a gel with G' > 10 kPa. This material was also self-healing and could recover its stiffness after large shear (Figure 3C). These gels’ stiffness was tunable, and could span the range of brain tissue and brain cancer stiffnesses20. It was observed that the same concentration of BSA with intact tertiary structure did not result in gelation (Figure 3E). This is perhaps owed to the fact that in its folded form, BSA protein exhibits singular charged binding pocket that cannot facilitate multichain interactions (Figure 1B), and thus cannot serve as an effective crosslinker. Partially denatured BSA, however, with multiple exposed charged peptide domains shows greater affinity towards multi-chain electrostatic interactions and crosslinks the system to form a physical gel. The difference in structure between these systems can be seen further in scanning electron microscopy (SEM) images (Figure 3F-H). Partially denaturing BSA results in some transition from a single binding pocket to the ability to interact with multiple chains, likely via electrostatic interactions. This transition may be due to changes in protein conformation making glycan binding sites more accessible, or aggregation of BSA from partial unfolding of the polypeptide21. This simple, electrostatic-based gelation is analogous to alginate gelation with divalent cations22. In this case, partially denatured proteins allow for bridged flocculation-type interactions which percolates a physical, non-covalent network. This is further reinforced by the shear-recovery observed in Figure 3C. Our group recently explored the differences in effect of physical or covalent bonds in energy transfer to physical or covalent networks23, and this shear-recovery is consistent with a physical network that can disassemble with shear and recover in situ over time.
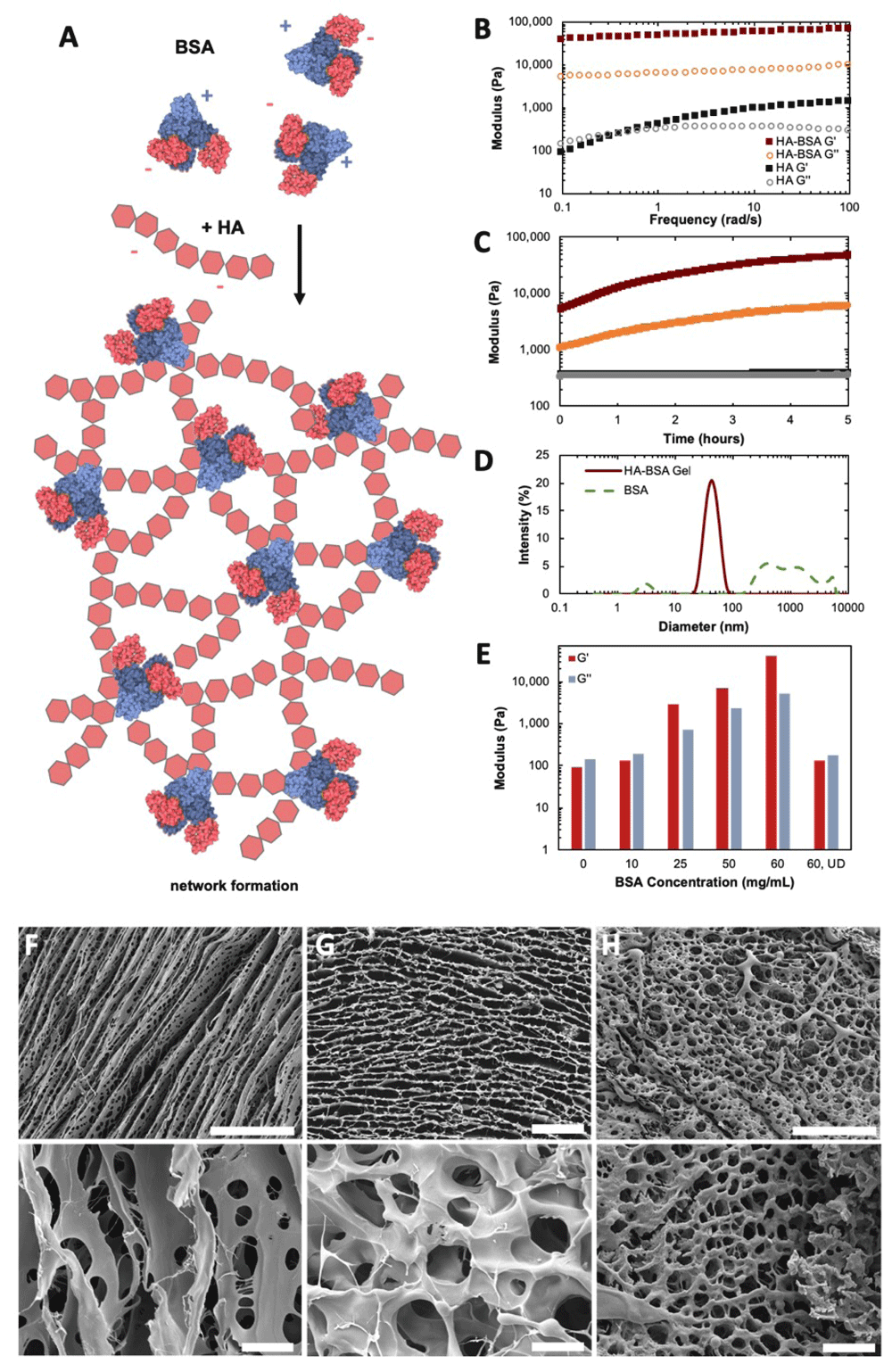
(A) Illustration of dynamic network formation of HA and BSA. (B) Oscillatory rheological frequency sweep of HA solution alone (40 mg/mL) and HA/BSA gels (40, 60 mg/mL). (C) Oscillatory time sweep after 100% shear of these HA/BSA gels and HA solution alone. Maroon = HA/BSA G’, Gold = HA/BSA G”, Black = HA G’, Grey = HA G”. (D) DLS size measurements of dynamic BSA particles alone and this HA/BSA gel. (E) Storage (G’) and loss (G”) moduli of HA (40 mg/mL) with various concentrations of BSA. UD = undenatured. (F–H) SEM images of HA alone, HA/BSA (undenatured), and HA/BSA (partially denatured). Scale bar = 100 µm (top), 10 µm (bottom). Dataset 1: Rheology, dynamic light scattering, scanning electron microscopy data.
Upon introduction of dry CS into a HA/BSA matrix, a sharp decrease in stiffness was observed (Figure 4A). We hypothesize this effect is due to interference from CS/BSA interactions leading to a reduction in extent of HA crosslinking. DLS data showed that when combined with either HA or CS, BSA exhibited hierarchical assemblies that were not observed for BSA alone. Furthermore, when combined with HA, the BSA assemblies were more dynamic than with CS (Figure 3D, Figure 4B, Figure 5). These measurements qualitatively suggest that BSA aggregates are not static and are polydisperse, and do not suggest that there are discrete BSA particles within the HA/BSA hydrogel. When all components are combined, there appears to be an intermediate solution that is pushed towards more well defined assembles immediately after shear but over time will revert towards the HA/BSA state (Figure 4B).
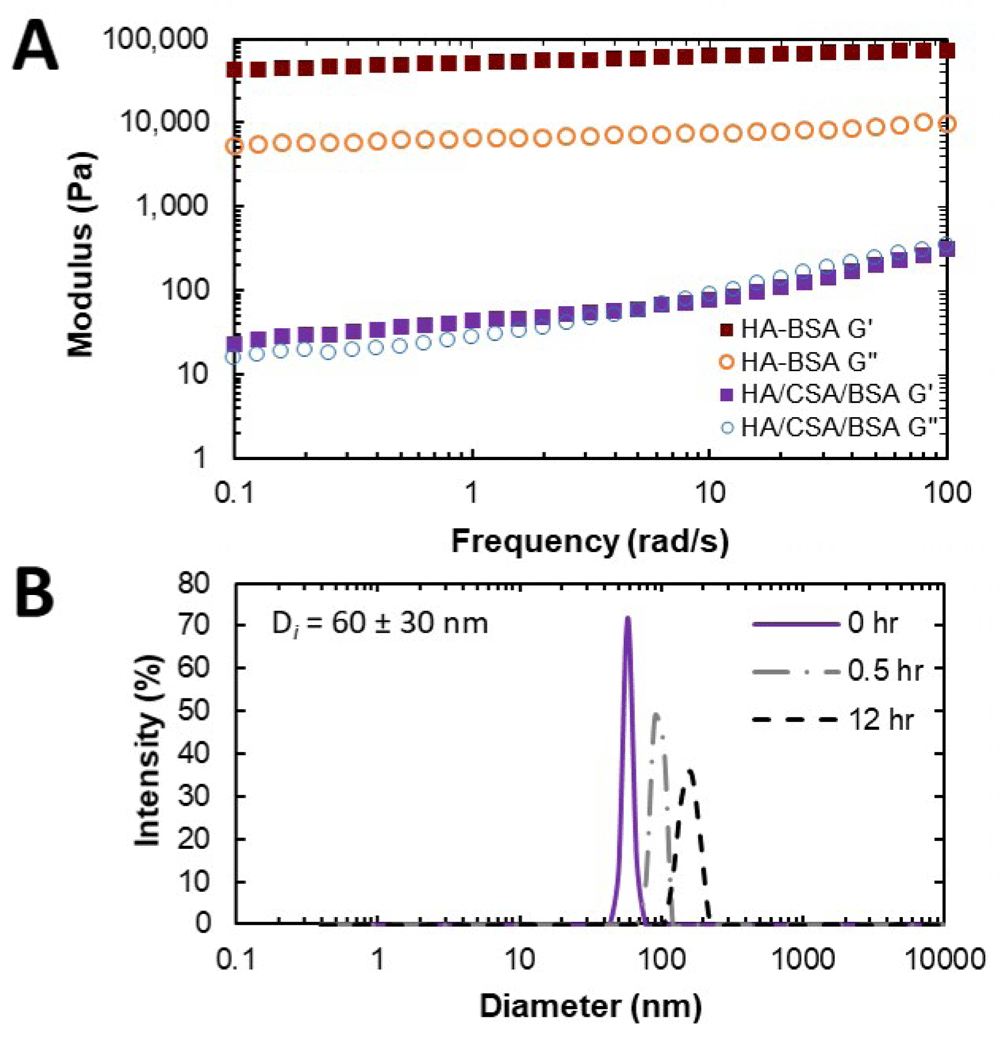
(A) Oscillatory frequency sweeps of HA systems loaded with BSA and with or without CSA. (B) Time-resolved dynamic light scattering experiment of CSA/BSA particles loaded with HA and sheared. Dataset 1: Rheology and dynamic light scattering data.
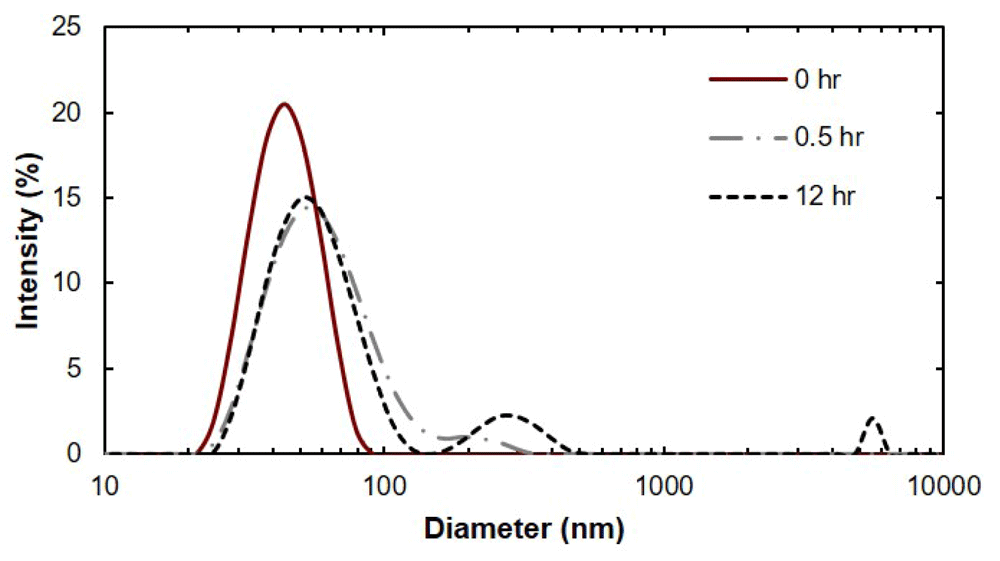
Dataset 1: Dynamic light scattering data.
We then explored whether these HA/CS/BSA systems could be used to study transport phenomena of the model hydrophilic drug rhodamine B (rhodB). Many parenteral drug-delivery studies monitor in vitro release kinetics into saline as a model for in vivo release. Here we explore the potential for a new ECM-mimetic model, in place of saline, to monitoring diffusion across a tissue-like gel (Figure 6). A hydrophobic blend of poly(caprolactone) (PCL) chains at different molecular weights (ESI) loaded with rhodB was carefully added on top of the hydrogel phase. We found that it was possible to monitor the interfacial concentration of rhodB in the hydrogel phase with UV-Vis spectroscopy. Interestingly, we observed a large bolus release of the hydrophilic drug from the hydrophobic phase to the hydrophilic phase at the interface until the same concentration was reached, subsequently an equilibrium or pseudo-steady-state concentration was reached after 14 h. Such a system is potentially useful in modeling mass transfer of drugs between hydrophobic drug delivery materials and biological tissue such as the brain24. Finally, prior tissue engineering studies have largely neglected the question of how the exact composition of the brain ECM might inform efforts to create biologically mimetic hydrogels. To estimate the physiological concentrations of CS and HA that occur in vivo (Figure 7, Dataset 117), we summarize the literature for CS and HA measurements of mammalian brain tissue25–32. These estimates clustered in the millimolar range for CS disaccharides, and 100 micromolar range for HA.
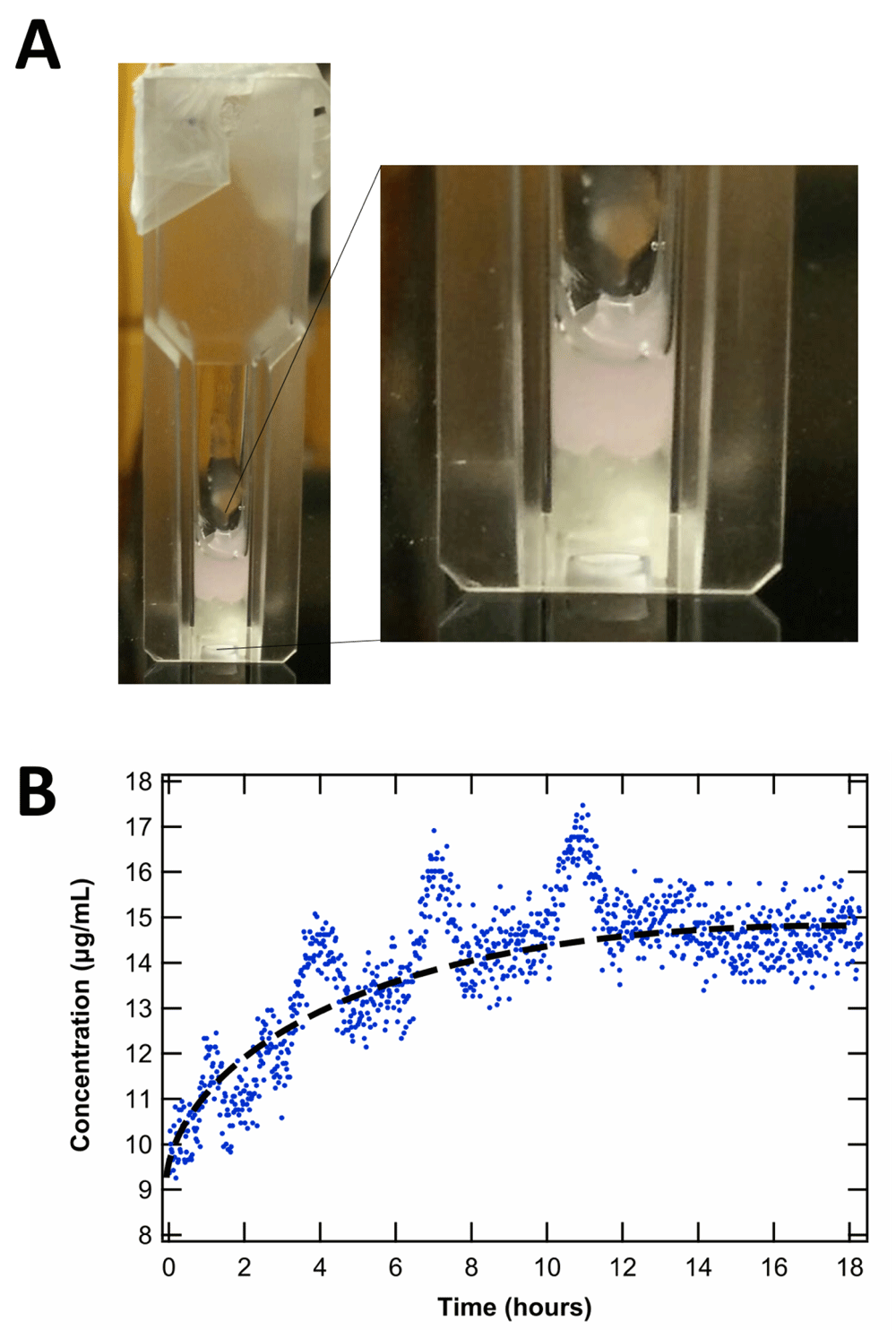
(A) Picture of PCL loaded with rhodamine (pink) on top of the HA/CS/BSA system, representing the hydrophobic and hydrophilic interface for potential modeling of mass transfer in drug delivery via UV-Vis spectroscopy. (B) Concentration at interface after large bolus release over time. Hydrophobic phase drug concentration = 10 µg/mL. Average hydrophilic phase equilibrium concentration = 14.5 µg/mL. The periodic fluctuations in concentration were attributed to trapped air interfering with the interface. (Dataset 1: UV-Vis data.
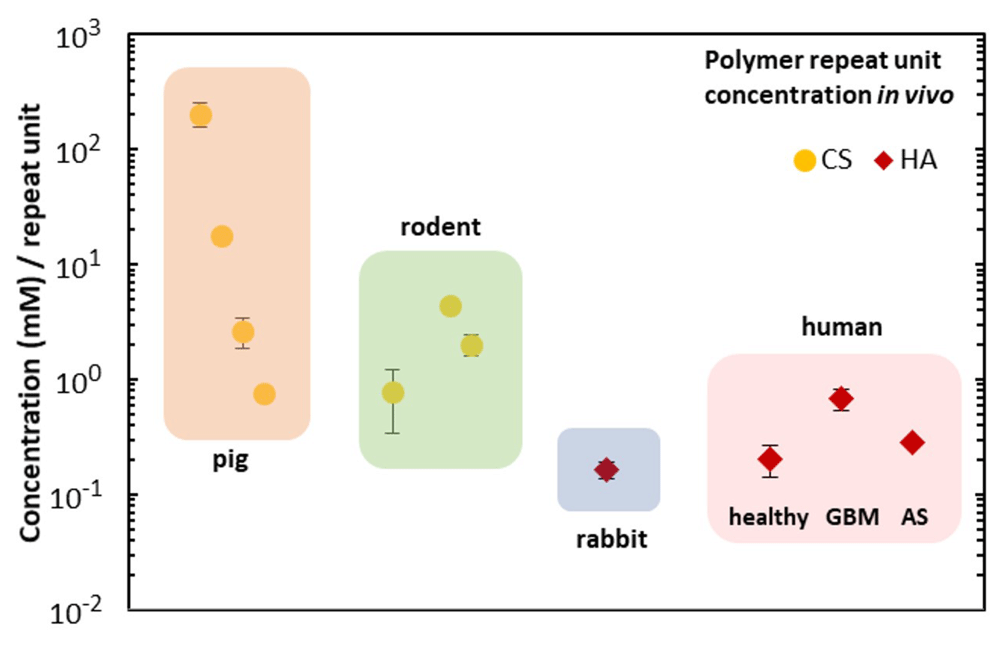
The concentration of HA in healthy tissue, glioblastoma, and astrocytoma is also plotted.
In this work we characterize non-covalent interactions between HA, CS, and BSA. We report nano- and microparticle self-assembly as well as gelation and supramolecular network formation via bridged flocculation. We also report on the mass transfer of a model drug from a hydrophobic phase into a glycosaminoglycans-based hydrogel, and suggest this approach may be more biologically representative of in vivo drug delivery compared to in vitro experiments that measure drug release from a matrix into saline. Finally, we summarize reported concentrations of CNS HA and CS in different animal models and humans, which may be important in the design of matrices for CNS tissue engineering. This work offers a robust method of forming hydrogels from unfunctionalized HA, and is attractive both for its simplicity and for capturing more biologically relevant interactions compared to those derived from chemically functionalizing either polysaccharide.
Underlying data for this study is available from Open Science Framework
OSF: Dataset 1: Protein-mediated gelation and nanoscale assembly of unfunctionalized hyaluronic acid and chondroitin sulfate, https://doi.org/10.17605/OSF.IO/3BXQG17
Data is available under a CC0 1.0 License
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)