Keywords
Mitochondria, mitochondrial Ca2+ uptake, mitochondrial ion transport, MCU, MICU
Mitochondria, mitochondrial Ca2+ uptake, mitochondrial ion transport, MCU, MICU
Ca2+ is universally recognised as one of the most pleiotropic second messengers in cell biology. Indeed, Ca2+ ions are responsible for decoding a variety of extracellular and intracellular stimuli, which, in animals, range from endocrine secretion to gene expression, muscle contraction, and synaptic transmission1–5. The efficacy of Ca2+ as a signalling molecule relies mainly on the maintenance of a steep Ca2+ gradient between the concentration in the extracellular (few mM) and intracellular (∼100 nM) environments. This >10,000-fold difference ensures that even very small changes in intracellular Ca2+ concentration ([Ca2+]i) are effective in regulating the numerous Ca2+-sensitive proteins of the cell, such as catalytic enzymes, channels, and transcription factors6. The maintenance of a low [Ca2+]i is tightly controlled by the presence of pumps and transporters both at the plasma membrane7 and at the membrane of organelles accumulating large amounts of Ca2+ 8,9. The endoplasmic/sarcoplasmic reticulum (ER or SR in striated muscle) is undoubtedly the main intracellular Ca2+ store; nevertheless, other cellular compartments actively participate in modulating [Ca2+]i: first of all mitochondria but also the Golgi apparatus10, endosomes, and lysosomes2,5,11.
The role for mitochondria in Ca2+ handling was first demonstrated in the 1960’s, when their ability to actively accumulate Ca2+ was evaluated in different cellular ex vivo models12–14. This occurred even before the advent of the chemiosmotic theory15, which provided the thermodynamic basis for Ca2+ entry into mitochondria. In addition, the role for mitochondria in cell function has gradually expanded, and its initial identification as the powerhouse for cellular energy supply was subsequently integrated into a more complex activity that includes the regulation of cell death, metabolism, and signalling pathways16–18. Notably, many mitochondrial functions are directly regulated by the level of Ca2+ ions inside the organelles4,19. The control of mitochondrial Ca2+ concentration ([Ca2+]mt) is thus of primary relevance for cell physiology, and emerging evidence converges on the concept that its dysregulation is of utmost importance in the establishment of pathological conditions.
In this commentary, we will focus on the molecular machinery underlying mitochondrial Ca2+ uptake, the mitochondrial Ca2+ uniporter (MCU) complex (see Figure 1), to which our laboratory has dedicated much effort in the last decade, contributing to its discovery in 2011 and unravelling its molecular complexity and functional significance.
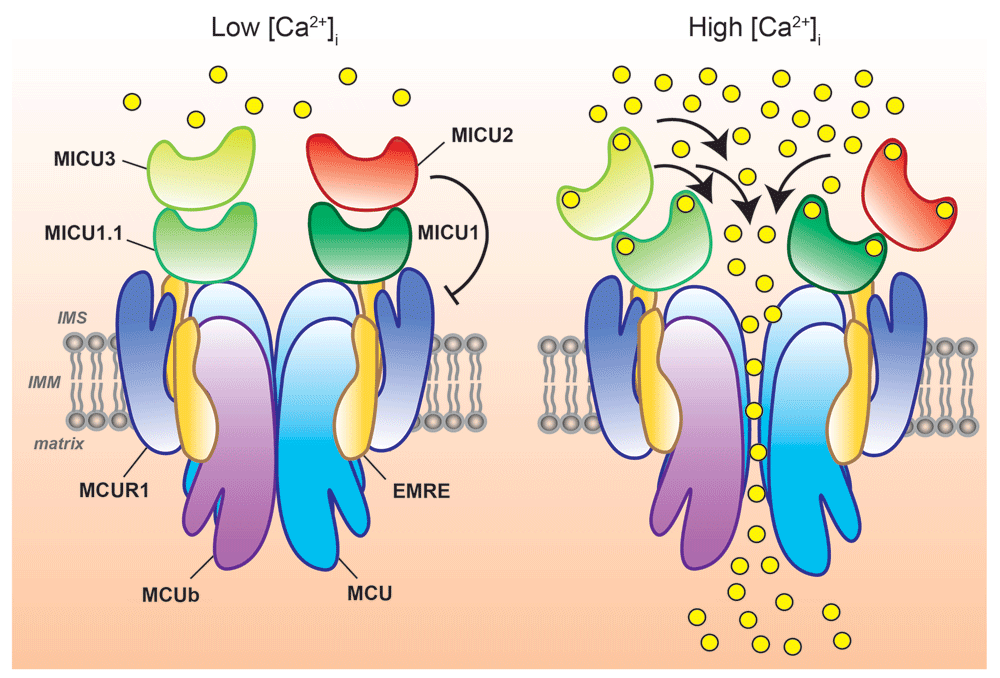
Schematic representation of MCU-mediated Ca2+ entry into mitochondria at different intracellular Ca2+ concentrations ([Ca2+]i). Mitochondrial Ca2+ uptake is controlled by a multiprotein complex consisting of MCU and MCUb (the pore-forming subunits) together with the essential mitochondrial Ca2+ uniporter regulator (EMRE), the mitochondrial Ca2+ uptake (MICU) proteins, MICU1, MICU1.1, MICU2, and MICU3, and, possibly, the MCU regulator 1 (MCUR1). At low [Ca2+]i, MICU1/MICU1.1–MICU2 or MICU1/MICU1.1–MICU3 heterodimers ensure MCU gatekeeper activity, preventing undesirable mitochondrial Ca2+ cycling in resting cells. At high [Ca2+]i, the MICU proteins act as positive regulators of MCU channel activity, allowing efficient mitochondrial Ca2+ uptake (right). IMS, intermembrane space; IMM, inner mitochondrial membrane.
Since the first reports of mitochondrial Ca2+ uptake12–14, extensive studies have been conducted to identify the mitochondrial Ca2+ entry mechanism, characterised by Ruthenium Red sensitivity, high selectivity, and high capacity for the cation20. Only from 2010, with the identification of the first regulator of the mitochondrial Ca2+ channel, the mitochondrial Ca2+ uptake protein 1 (MICU1)21, and, one year after, by the cloning and molecular characterisation of the gene coding for the MCU22,23, the entry pathway was characterised at the molecular level and the route for the genetic manipulation of mitochondrial Ca2+ uptake was finally open.
In 2011, two independent in silico screenings22,23 identified the CCDC109a gene product as the long-sought pore-forming unit of the MCU channel mediating mitochondrial Ca2+ uptake in mammalian cells. Purified MCU protein reconstitution in lipid bilayers revealed a Ca2+ channel activity with the electrophysiological features of the hypothetical MCU20. In addition, it was clearly shown that MCU downregulation in mammalian cells inhibits mitochondrial Ca2+ uptake while its overexpression increases mitochondrial Ca2+ accumulation, thus proving that it is bona fide the channel mediating Ca2+ uptake in mitochondria23.
The sequence of MCU consists of two transmembrane α-helix domains spanning the inner mitochondrial membrane (IMM), the second of which contains the critical DIME motif responsible for the channel selectivity filter, linked by a short loop toward the mitochondrial intermembrane space (IMS), while both the C- and the N-termini face the mitochondrial matrix22,24. A single MCU molecule per se is thus not able to form the channel but should organise in oligomers, and MCU was initially proposed to arrange itself into tetramers25. This hypothesis was very recently confirmed by coherent high-resolution cryo-EM data from four independent groups on different MCU orthologs24,26–28. These seminal studies finally unveiled the structure and arrangement of MCU protomers within the complex and the exact position of the channel selectivity filter. The 3D reconstruction of EM data unambiguously revealed that MCU forms tetramers with a nonobvious symmetry: the transmembrane domain displays a fourfold symmetry, while the N-terminal domain (NTD) on the matrix side shows a twofold symmetry axis. These findings confuted the pentameric MCU architecture that was previously proposed based on magnetic resonance and negative staining EM data from Caenorhabditis elegans MCU-ΔNTD protein, in which a significant portion of the NTD was removed to facilitate the analysis29. In addition, the MCU DIME motif, which contains the two critical acidic residues directly involved in the cation coordination, has now been shown to reside at the beginning of––and thus integral to––the second transmembrane helix (TMH), and not in the loop connecting the two transmembrane helices, as previously suggested29.
However, as Raffaello et al. showed, MCU is not the only pore-forming unit of the oligomer, but it associates with the protein encoded by the MCU paralog gene CCDC109b, which was thus named MCUb25. The characterisation of MCUb demonstrated that it acts as a negative regulator of MCU activity both in lipid bilayer experiments and when overexpressed in mammalian cells25 (see Figure 1). To note, in other organisms, such as Trypanosoma cruzi, the ortholog of MCUb acts as a Ca2+ conducting subunit and its overexpression enhances, rather than dampens, mitochondrial Ca2+ uptake30. Interestingly, in another trypanosomatid, Trypanosoma brucei, two additional MCU isoforms were identified, MCUc and MCUd, which are also endowed with Ca2+ uniporter activity and are able to form heterotetrameric complexes with their homologues MCU and MCUb31. This highlights the existence of significant species-specific differences in the function and distribution of MCU orthologues, despite their sequence conservation, and this should be taken into account when studying different organisms. Moreover, the expression and relative proportion of MCU and MCUb vary significantly among different tissues in mammals; thus, each cell type may have different variants of the uniporter owing to the specific composition and stoichiometry of MCU subunits. This accounts for tissue-specific variations of the capacity of mitochondria to take up Ca2+ and perfectly fits with the electrophysiological recordings of MCU Ca2+ currents in mitochondria from different mammalian tissues32. In skeletal muscle, for example, the presence of a high MCU:MCUb ratio25 matches the highest mitochondrial Ca2+ conductance recorded in this tissue32, whereas, in adult heart, the relatively elevated MCUb expression25 results in a considerably low Ca2+ current in cardiomyocyte mitochondria32. In cardiac cells, in which ∼37% of the volume is occupied by mitochondria33, this is crucial for preventing massive mitochondrial Ca2+ accumulation that would potentially cause undesired buffering of the [Ca2+]i transients required for contraction, futile cycling of Ca2+ across the IMM, and, eventually, organelle Ca2+ overload and apoptosis. The control of the MCU:MCUb proportion in the mitochondrial Ca2+ channel pore domain is thus fundamental for the function and physiology of different tissues.
After the molecular identification of the MCU pore components and auxiliary factors, the path for the genetic manipulation of mitochondrial Ca2+ uptake machinery in cells and animal models was finally opened. The first MCU–/– mouse was generated by Finkel’s group in 2013. It shows a relatively mild phenotype, and, despite the expected abrogation of mitochondrial Ca2+ uptake, it develops normally and displays unaffected basal metabolism34. However, the MCU–/– mouse shows increased plasma lactate levels after starvation and impaired exercise performance accompanied by a reduction in the activity of pyruvate dehydrogenase in skeletal muscle34. These findings, together with the fact that MCU–/– mouse survival depends on the genetic background (MCU deletion is embryonically lethal in a pure C57/BL6 background35), pointed to a more subtle and less obvious involvement of mitochondrial Ca2+ uptake in organ metabolism and organism development. This issue has been elegantly addressed by recent studies implementing tissue-specific modulation of MCU expression (by ablation/downregulation or overexpression) in adult tissues. For example, cardiac-specific tamoxifen-inducible MCU deletion in adult mice, differently from the germline genetic ablation of MCU, which did not protect from ischemic-reperfusion injury34, clearly protects from ischemia-reperfusion heart damage, abrogates the contractile responsiveness to β-adrenergic stimulation responsible for the so-called “fight-or-flight” response, and reduces heart bioenergetics reserve capacity, even though it does not induce phenotypic abnormalities either in basal conditions or after cardiac overload and does not alter cardiomyocytes’ resting [Ca2+]mt36–39. These results suggest that MCU may be dispensable for cardiac homeostasis in basal conditions, while it appears to play a major role in cardiac metabolic flexibility during acute stress. Similarly to what has been reported for the heart tissue, germline deletion of MCU did not protect from hypoxic/ischemic brain injury40. However, brains from conditional neuronal-specific inducible MCU-deleted mice as well as primary cortical neurons silenced for MCU show a significant reduction of the hypoxic/ischemic damage and decreased cell death without impairment of neuronal mitochondria metabolism41. Once more, these findings point to the existence of a strong drive for organs and tissues to respond to chronic MCU ablation by establishing adaptive metabolic shunts in order to cope with/bypass impaired mitochondrial Ca2+ signalling. Such adaptation to mitochondria dysfunction has also been described in the cardiac-specific Tfam–/– mouse, the murine model for human cardiomyopathies with mtDNA depletion42, in which the alteration in mitochondrial metabolism due to oxidative phosphorylation (OXPHOS) deficiency induces a secondary upregulation of MCU protein and a concomitant downregulation of NCLX transcription43. The resulting increased mitochondrial Ca2+ uptake and reduced Ca2+ efflux in Tfam–/– cardiac mitochondria, although inducing an elevated and potentially damaging [Ca2+]mt, appear somehow functional to ensure an efficient Ca2+-dependent mitochondrial respiration that otherwise would be compromised by the pathology41.
Overall, these findings lead us to the following consideration: experimental systems featuring long-term genetic manipulation of the mitochondrial Ca2+ uptake machinery should be evaluated with caution, and the potential induction of unpredicted phenotypes, as a consequence of adaptive/maladaptive responses, should be taken into account. This is particularly relevant when in vivo models are considered.
In line with this, the acute but transient manipulation of MCU expression in skeletal muscle provided additional intriguing evidence of the wide-ranging influence of mitochondrial Ca2+ signalling in the maintenance of organ and organism homeostasis. The decrease or enhancement of [Ca2+]mt by AAV-mediated MCU silencing or overexpression, respectively, demonstrated that mitochondrial Ca2+ uptake regulates myofibre trophism through the modulation of the activity of the insulin growth factor-1/Akt pathway and the transcription of the peroxisome proliferator-activated receptor gamma coactivator 1 alpha 4 gene (PGC1α4). In particular, the overexpression of MCU in both adult and neonatal muscles induces significant fibre hypertrophy, which is not accompanied by alterations in mitochondrial membrane potential or other metabolic parameters (such as glycogen content and succinate dehydrogenase activity)44. On the contrary, MCU downregulation leads to marked fibre atrophy as demonstrated by reduction of fibre size, inhibition of Akt activity, reduction of PGC1α4 transcripts, and impaired activation of the pyruvate dehydrogenase complex44, in agreement with the data from the MCU–/– mice34 (see Figure 2).
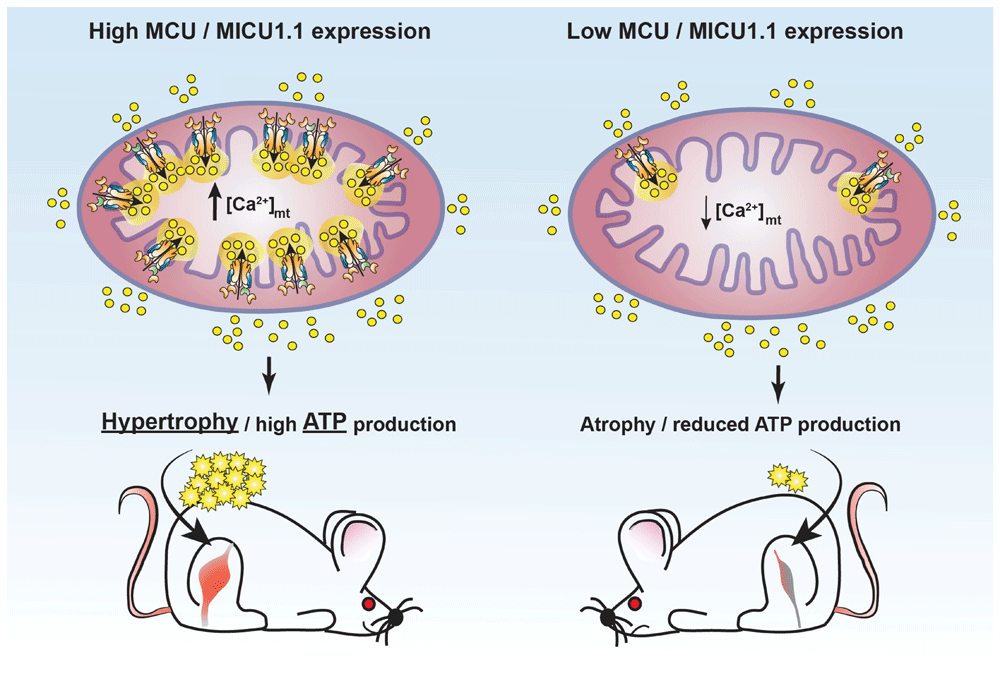
Schematic representation of the effects of manipulating mitochondrial Ca2+ uptake in skeletal muscle fibres. Increased mitochondrial Ca2+ accumulation by enhanced expression of mitochondrial Ca2+ uniporter (MCU) or mitochondrial Ca2+ uptake protein 1.1 (MICU1.1) leads to muscle hypertrophy by stimulating fibre growth and ATP production (left). Reduction of mitochondrial Ca2+ uptake by downregulation of either MCU or MICU1.1 causes fibre atrophy and impaired ATP production (right). [Ca2+]mt, mitochondrial Ca2+ concentration.
Moreover, the recent description of a mouse model bearing constitutive skeletal muscle-specific deletion of MCU (skMCU–/– mice) has been fundamental to uncover the metabolic rewiring occurring at the level of skeletal muscle cell but also, importantly, systemically45. In line with previous data34,44, the muscle of skMCU–/– mice shows decreased performance and smaller fibre size compared to that of control mice45. Moreover, it shows a shift toward fast fibre type and an enhanced preference for fatty acid oxidative metabolism45. Interestingly, the ablation of MCU in skeletal muscle also impinges on other tissues, as shown by the increased catabolic response in both liver and adipose tissue of skMCU–/– animals45. This clearly points out how the mitochondrial metabolic adaptation in response to altered Ca2+ dynamics (i.e. MCU deletion) within the muscle tissue may have major systemic impact on different tissues and, importantly, on the modulation of global metabolism.
MICU1 was the first regulator of mitochondrial Ca2+ uptake to be identified21, even before the identification of MCU itself. It was initially described as an EF-hand Ca2+-binding protein associated with the IMM that stimulates MCU channel opening and thus acts as a positive modulator of mitochondrial Ca2+ uptake. Subsequent findings defined the crucial role of MICU1 in keeping the channel inactive under resting conditions, i.e. at low [Ca2+]i46,47, thus preventing massive Ca2+ entry that will otherwise cross the IMM down the steep electrochemical gradient and cause harmful mitochondrial Ca2+ overload. MICU1 was thus appropriately defined as the gatekeeper of the MCU channel. This concept has been recently confirmed by two independent in vivo MICU1–/– mouse models. These MICU1–/– animals, despite normal embryonic development and growth, show a variable degree of postnatal lethality (one out of six to one out of seven surviving pups in the case of CRISPR/Cas9 mutants48 and 100% lethality in the case of Cre recombinase/LoxP deleted alleles49) mostly due to breathing failure for the underdevelopment of the respiration coordination centres (cerebellum and Purkinje cells)49. The few surviving knockout mice display muscle impairment and ataxia48, a phenotype very similar to that described in children with a MICU1 mutation50. The analysis of MICU1–/– mitochondria revealed increased matrix [Ca2+]mt, increased sensitivity to permeability transition pore (PTP) opening and cell death, and reduced production of ATP, similar to what was reported in MICU1-silenced hepatocytes and Hela cells46,47. Overall, these data clearly highlight the relevance of MICU1 gatekeeping function in the control of mitochondrial Ca2+ homeostasis and metabolism. Nevertheless, an additional intriguing feature of MICU1 action has recently emerged, which consists of its ability to confer MCU selectivity for Ca2+ over Mn2+. Indeed, the DIME motif in the MCU pore subunit appears to be unable per se to discriminate between Ca2+ and Mn2+ in the absence of MICU151. However, when present, MICU1 prevents Mn2+ entry in mitochondria, thus ensuring the stringent Ca2+ selectivity of MCU51. In this scenario, the proper stoichiometry of MICU1 is thus crucial to avoid mitochondrial Mn2+ uptake and to prevent Mn2+ toxicity. This concept may have relevant implications for human pathologies, especially for those diseases caused by MICU1 deficiency50,52,53 but also for Parkinson’s disease, as already suggested51.
Additional MICU family members have been identified by genome sequence analysis soon after MICU1, namely MICU2 (EFHA1) and MICU3 (EFHA2)54, which share 41% and 34% identity with MICU1, respectively, but display a broad and non-overlapping expression pattern compared to MICU155. In particular, MICU2 was shown to bind covalently to MICU1 through disulphide bridges, and the resulting dimer endows the genuine MCU gatekeeper activity56. In this scenario, cytosolic Ca2+ elevation causes Ca2+ binding to the EF-hand domains of the MICU1–MICU2 dimer, allowing MICU1 to function as a positive regulator of MCU activity, thus efficiently promoting Ca2+ entry into the organelle56 (see Figure 1).
As for MICU3, its evolutionary conservation and sequence similarity to MICU1 suggested that it may share a common biological function with MICU154. Along this line, its role as a positive regulator of mitochondrial Ca2+ uptake has been demonstrated recently55. Indeed, the expression of MICU3 in Hela cells, which normally do not express it, showed that it interacts with MICU1, but not with MICU2, forming obligatory dimers55 (see Figure 1). This interaction causes a significant increase of mitochondrial Ca2+ uptake, demonstrating the stimulatory action of MICU3 on MCU activity. In addition, while displaying a reduced gatekeeper activity, MICU3 has been shown to mediate a more rapid response of mitochondria to [Ca2+]i changes, allowing a shorter delay between the [Ca2+]i rise and the increase in mitochondria Ca2+ uptake compared to MICU155. MICU3 thus plays a pivotal role in determining the kinetics of mitochondrial Ca2+ signalling and consequently in the modulation of global [Ca2+]i dynamics, which may be of utmost relevance for the decoding of Ca2+ signals in excitable cells, such as neurons, where indeed MICU3 shows the highest level of expression54,55.
By the interaction of monomers with non-overlapping functions, the MICU proteins confer an important property of the mitochondrial Ca2+ uptake system: the sigmoidicity of the dose-response to Ca2+. Indeed, the activity of MCU, which is kept low at resting [Ca2+]i (∼100 nM) owing to the MICU1–MICU2 gatekeeper action, as already discussed, becomes extremely efficient upon [Ca2+]i elevation. Indeed, Ca2+ binding to the MICU1–MICU2 regulatory complex converts it from an inhibitory regulator (ensuring gatekeeping) at basal [Ca2+]i to an activator, strongly enhancing the flux through the channel. The threshold for MCU activation is directly dependent on the Ca2+ affinity of MICUs’ EF hands (which range from 300 nM to 1.3 µM), since the cooperative Ca2+ binding to these domains induces a conformational change in the MICU1–MICU2 dimer, which acts as a molecular switch relieving MCU inhibition57.
Despite the role for MICU2 in the regulation of MCU gatekeeping and activation being controversial still, an intriguing hypothesis is that it has evolved to spatially restrict the Ca2+ crosstalk between single inositol trisphosphate receptor (IP3R) and MCU channels58. Indeed, the presence of MICU2 has been shown to decrease the Ca2+ affinity of the MICU1 gatekeeper, thus elevating the threshold for MCU opening58. In the context of the intracellular environment, this increased threshold for mitochondrial Ca2+ uptake translates into the need for a shorter distance between the ER Ca2+-releasing channel (IP3R) and the mitochondrial Ca2+ uptake channel (MCU)58.
In line with this, in the last few years, the stoichiometry of MCU components has emerged as a crucial feature of the modulation of mitochondrial Ca2+ uptake rate. The relative proportion of the MICU proteins, in particular, has been demonstrated as the basis of the tissue-specific differences in cellular Ca2+ dynamics59. Indeed, the different ratio of MICU1:MCU and MICU1:MICU2 proteins in the liver, the heart, and skeletal muscle has been correlated with the distinct mitochondrial Ca2+ handling of these three tissues59. The level of MICU1 expression, which is much lower in cardiac tissue (while MCU and MICU2 are present at similar levels) compared to liver, has been suggested to determine the lower maximal Ca2+ uptake capacity and less-steep Ca2+ dependence that characterise cardiac mitochondria compared to liver mitochondria, as electrophysiological data32 and intracellular Ca2+ measurements59 revealed. The low amount of MICU1 in cardiac mitochondria is instrumental to reduce mitochondrial Ca2+ uptake upon cytosolic Ca2+ elevation, allowing the decoding of the repetitive cytosolic Ca2+ spikes of the beating heart into a graded increase of matrix Ca2+. This integration of frequency fluctuations is crucial to prevent mitochondrial Ca2+ overload, which will be otherwise detrimental to the cardiac myocytes. By contrast, the relatively high MICU1 level and its cooperative activation of MCU in hepatocytes ensure the punctual and massive mitochondrial Ca2+ increase at every single cytosolic Ca2+ spike59, which is functional to liver oxidative metabolism.
An additional degree of complexity came with the discovery of an alternative MICU1 isoform, extremely conserved and expressed at the highest level in skeletal muscle, which originates from the alternative splicing of the MICU1 mRNA, the MICU1.1 splice variant60 (see Figure 1). This alternative isoform was shown to dimerise with MICU2, showing gatekeeper activity similar to that of MICU1, but it binds Ca2+ one order of magnitude more efficiently than MICU1 and activates MCU at lower [Ca2+]i than MICU160. Thus, in skeletal muscle, the MICU1.1–MICU2 dimer ensures a much higher net Ca2+ entry and elevated ATP production than the MICU1–MICU2 dimer60. These results highlight a novel mechanism of the molecular plasticity of the MCU Ca2+ uptake machinery that allows skeletal muscle mitochondria to adapt to the intense metabolic challenge that characterises this tissue (see Figure 2).
To add further complexity to the study of MCU channel regulation, other molecules have been shown to interact with MCU in recent years. Indeed, the quantitative mass spectrometry analysis of the MCU-interacting proteins revealed the presence of a critical component of the MCU complex, the essential MCU regulator (EMRE) (see Figure 1), a 10 kDa single-pass transmembrane protein that was proposed to be necessary for MCU function by bridging MCU interaction with MICU1 in mammalian cells61. Interestingly, EMRE appears to be required for MCU channel activity in metazoans only, since it is not found in plants, fungi, or protozoa, such as Dictyostelium discoideum, although they express MCU proteins with functional channel activity. While its exact role in the MCU complex is still uncertain, EMRE was proposed to mediate MCU sensitivity to matrix [Ca2+]mt, suggesting that MCU activity may be modulated by Ca2+ sensors facing both the IMS (MICUs) and the matrix (EMRE)62. However, this hypothesis was subsequently questioned by other groups, whose data supported an opposite topology for the EMRE protein, with the short N-terminus exposed to the matrix while its acidic C-terminus faces the IMS63,64. In this conformation, EMRE action would be that of supporting MCU Ca2+ transport activity by interacting with the first TMH of MCU within the IMM and to interact also with MICU1 at the IMS via its C-terminus poly-aspartate tail, thus ensuring MICU1 binding to the channel and gatekeeping activity63.
Recently, the control of EMRE protein levels has emerged as a crucial aspect of MCU complex regulation. Indeed, a correct amount of EMRE is fundamental to guarantee the exact stoichiometry of the different MCU components. Alteration of EMRE protein levels by ablation of the AAA-proteases responsible for its degradation65, or by expression of proteolytic-resistant EMRE constructs62, leads to uncontrolled MCU channel opening causing mitochondrial Ca2+ leakage and mitochondrial Ca2+ overload, resulting in neuronal cell death65,66. The existence of a tight control of EMRE protein levels has also been demonstrated in vivo in the MICU1–/– mice, where EMRE is reduced in a genetic condition characterised by increased [Ca2+]mt basal levels48. These findings clearly underline a negative regulatory mechanism that keeps EMRE expression in check in order to cope with changes in MCU activity48.
Another protein that has been associated with the MCU complex67,68, despite the original proteomic analysis of the MCU interactors failed to recover it61, is the MCU regulator 1 (MCUR1). MCUR1 is a coiled-coil-containing protein encoded by the CCDC90A gene initially identified by a RNAi screen searching for a mitochondrial membrane protein involved in MCU homeostasis67, whose precise function in the regulation of MCU activity and mitochondrial metabolism is still debated. Indeed, MCUR1 has been shown to be required for MCU-dependent Ca2+ uptake, since its knockdown dampens mitochondrial Ca2+ entry while its overexpression enhances it67. However, a direct action of MCUR1 on MCU activity was questioned by the finding that its silencing leads to a significant loss of mitochondrial membrane potential (ΔΨm), which per se can account for the decreased mitochondrial Ca2+ uptake69. In this context, MCUR1 has been suggested to act primarily as a cytochrome c oxidase (COX) assembly factor69, and this concept would also be supported by the fact that a MCUR1 orthologue is present in budding yeast70, which lack MCU, and is required for yeast COX activity and survival in non-fermentable medium69. Nevertheless, the patch clamp data of Ca2+ currents from voltage-clamped mitoplasts point to an effective role for the MCUR1 protein on MCU channel conductance, independently of ΔΨm71. In line with this, experiments in Drosophila cells, which are resistant to Ca2+-induced mitochondrial permeability transition (MTP)72 and in which no MCUR1 homologs are found, show increased sensitivity to Ca2+-dependent PTP opening when heterologous human MCUR1 is expressed, which is not accompanied by alterations in the rate of mitochondrial Ca2+ uptake72. Conversely, MCUR1 knockdown in mammalian cells renders them resistant to Ca2+ overload, providing protection from cell death72. This led the authors to conclude that MCUR1 regulates the Ca2+ threshold for the MPT and may act as a molecular bridge connecting the MCU channel to the MTP complex. Whether this hypothesis holds true still has to be tested.
More recently, the genetic deletion of MCUR1 in endothelial and cardiac tissues in vivo by mouse conditional knockout models68 points to an involvement of MCUR1 in the assembly and function of the MCU channel. In particular, MCUR1 binding to MCU and EMRE appears to be necessary for MCU oligomerisation and stability, since MCUR1–/– cells will display a significantly decreased amount of MCU-containing high-molecular-weight complexes and reduced rate of mitochondrial Ca2+ uptake68. This, as expected, impinges on cellular energetics, as it is reflected by the dramatic reduction of ATP levels and consequent activation of the pro-survival autophagy pathway in cells depleted of MCUR172. Whether the pleiotropic roles for MCUR1 in the regulation of mitochondrial Ca2+ uptake, OXPHOS efficiency, and the apparent discordant phenotypes of MCUR1-depleted cells depend on the intrinsic transcriptional and metabolic differences of different cell types or on the species-specific features of the mitochondrial Ca2+ machinery is still an open question, and further investigation is needed to clarify it.
The regulation of the mitochondrial Ca2+ uptake machinery has been revealed to be far more complex than that resulting from just considering the cooperative actions of the different MCU components and their possible combinations. Indeed, the activity of the MCU channel relies on a multifaceted integration of regulatory mechanisms acting on both the MCU pore subunits and its co-regulators. Moreover, these mechanisms have been shown to occur at multiple levels––transcriptional, post-transcriptional, and post-translational––and their systematic identification has started to be accomplished only in recent years.
In neurons, for example, MCU transcription appears to be under the control of activity-dependent cytosolic Ca2+ signalling through the involvement of calmodulin (CaM) and the activation of CaM-kinase (CaMK)73. It has been shown that the immediate-early gene Npas4, a neuronal transcription factor with neuroprotective action against excitotoxicity74, acts directly downstream of the NMDAR signalling and of CaMK to modulate MCU transcription73. In another context, chromatin immunoprecipitation and promoter reporter analyses revealed that the Ca2+-regulated transcription factor cyclic adenosine monophosphate response element-binding protein (CREB) directly binds the MCU promoter and stimulates its transcriptional activity in chicken DT40 lymphocyte cells75. The CREB-mediated activity-dependent modulation of MCU expression in response to intracellular Ca2+ mobilisation via IP3R and store-operated Ca2+ entry (SOCE) has also been proven to be crucial to ensure a prompt metabolic flexibility and cell survival in lymphocytes75.
The existence of post-transcriptional regulation of MCU has been initially described in cancer76, where a strong inverse correlation between MCU expression level and the abundance of microRNA (miR)-25 has been reported in both tumour cell lines and colon adenocarcinoma samples76. Indeed, MCU expression is found to be upregulated in both colon and prostate cancer cells that show a reduced level of miR-25, and the overexpression of miR-25 in these cells downregulates MCU and increases cell sensitivity to apoptosis76. Similarly, in breast cancer, the downregulation of miR-340 is correlated with increased MCU expression in highly metastatic cells, while MCU targeting by miR-340 blocks the metabolic shift from OXPHOS to aerobic glycolysis that would otherwise favour cell migration and invasiveness77. In the context of pulmonary arterial hypertension (PAH), in which vascular cells are hyper-proliferative and apoptosis resistant, exhibiting a cancer-like phenotype, the decreased MCU function that underlies the key phenotypic features of PAH (including elevation of [Ca2+]i, reduction of [Ca2+]mt, mitochondrial fragmentation, and the Warburg phenomenon) has been ascribed to the downregulation of MCU expression by both miR-25 and miR-13878. The overexpression of MCU or the blocking of the MCU-targeting miRs in PAH cells indeed restores Ca2+ dynamics, mitochondrial metabolism, and cell proliferation to physiological levels78. MiR-25 has also been shown to target and downregulate MCU in the heart, where it is suggested to protect myocytes from oxidative damage by reducing [Ca2+]mt levels79. Another two muscle-specific miRNAs (myomiR), miR-1 and miR-206, have been implicated in the regulation of MCU expression in muscle80, of which miR-1 plays a major role in the context of cardiac development in both mice and humans. In mice, miR-1 upregulation is crucial during the first few postnatal weeks for the modulation of MCU activity in order to prevent massive [Ca2+]mt elevation that would otherwise occur, since mitochondria distribution undergoes heavy remodelling at this time point and organelles are placed in close contact with the SR Ca2+-releasing channels80.
Post-translational modifications of MCU proteins have also been described. Indeed, the Ca2+-dependent tyrosine kinase Pyk2 has been shown to directly interact and phosphorylate MCU in cardiac cells in response to α-adrenergic stimulation81. The MCU phosphorylation by Pyk2 has been proposed to promote its oligomerisation and to enhance mitochondrial Ca2+ entry, thus stimulating mitochondrial metabolism and favouring cytosolic Ca2+ clearance in cardiac cells81. In addition, the atomic resolution data of the N-terminus of MCU (residues 72–189) allowed the description of the exact structure of the MCU matrix domain82. This domain was shown to acquire a β-grasp-like fold and to bind Mg2+ and Ca2+ through a MCU-regulating acidic patch (MRAP)82. The disruption of cation binding via mutation of the MRAP sequence impairs MCU oligomerisation, reduces basal [Ca2+]mt levels, and leads to significant dampening of mitochondrial Ca2+ uptake after agonist stimulation, highlighting the role of divalent cations in the regulation of MCU activity81. This has been proposed as a possible feedback mechanism of MCU regulation aimed at preventing excessive mitochondrial Ca2+ entry in conditions of elevated [Ca2+]i.
Finally, a wide range of chemical modifications have been reported to target the thiol moiety of Cys residues in many proteins, enabling biological switching of structure and reactivity oxidation83. One of these modifications, S-glutathionylation, was recently shown to occur in MCU protein as the result of oxidation at the evolutionarily conserved Cys-9784. The S-glutathionylation has been demonstrated to promote a MCU conformational change, which increases oligomerisation84. In human pulmonary microvascular endothelial cells (HPMVECs), this enhances MCU activity and promotes increased mitochondrial Ca2+ entry both in basal conditions and upon agonist challenge, causing cellular bioenergetics crisis and higher sensitivity to cell death84.
Overall, the understanding of the mechanisms controlling Ca2+ entry in mitochondria has made a great step forward in the last few years, and new possibilities for effective modulation of MCU activity have been revealed. In this line, very recently, a systematic orthologous interspecies chemical screen based on the combination of an optimised yeast cell system and a mammalian mitochondria-based NCC library drug screen picked out and validated the first MCU-specific inhibitor molecule to be described: mitoxantrone85. Mitoxantrone has been utilised already in clinical practice for its antineoplastic action against non-Hodgkin’s lymphomas and acute myeloid leukaemia86; however, the anti-tumour properties of this drug appear to rely on a different molecular moiety with respect to its anti-MCU activity, thus opening up the possibility for the chemical engineering of new lead compounds to specifically target MCU function. This is of outmost relevance for the design of novel potential therapeutic approaches to pathologies in which mitochondrial Ca2+ signalling dysfunction is involved17,87,88, including those characterised by primary MCU dysfunction due to mutations in its components50,52,53.
In conclusion, despite many of the molecular mechanisms refining MCU activity in the cell having been described, of which the most relevant are presented in this review, a number of other possibilities for the modulation of uniporter function may exist (at transcriptional and post-transcriptional levels), and additional efforts are needed from the scientific community to fully unravel the complexity of mitochondrial Ca2+ uptake regulation.
In the last few years, tremendous advances have been made in defining the molecular identity of the mitochondrial Ca2+ transport machinery. Most of the components of the MCU complex have been identified, and several cellular and in vivo models have been generated that helped to define the physiological relevance of mitochondrial Ca2+ uptake. Still, many issues remain obscure and need to be investigated. The entire spectrum of molecular mechanisms by which different cell types finely tune mitochondrial Ca2+ uptake appears to be related not only to the metabolic needs but also to the breadth of cellular activities modulated by organelle Ca2+ levels. We believe that the complete picture will emerge when at least three conceptual mechanisms are fully investigated, i.e. gene expression (the MCU and MICU1 isoforms show significant tissue-specific differences in expression), alternative splicing (as in the case of MICU1.1), and post-transcriptional and post-translational regulation (which, so far, has been only partially explored). We then need to explore the functional interplay between mitochondrial Ca2+ transport and other ion fluxes, such as Na+ and K+, for which complexity and partial redundancy of transport mechanisms have been described but molecular definition still lags behind. Unravelling this cross-talk will greatly enhance our insight into the regulation of mitochondria, these fascinating organelles with pleiotropic effects on a cell’s life and death.
[Ca2+]i, intracellular Ca2+ concentration; [Ca2+]mt, mitochondrial Ca2+ concentration; CaM, calmodulin; CaMK, calmodulin kinase; CREB, cyclic adenosine monophosphate response element-binding protein; COX, cytochrome c oxidase; EMRE, essential mitochondrial Ca2+ uniporter regulator; ER, endoplasmic reticulum; IMM, inner mitochondrial membrane; IMS, intermembrane space; IP3R, inositol trisphosphate receptor; MCU, mitochondrial Ca2+ uniporter; MCUR1, mitochondrial Ca2+ uniporter regulator 1; MICU, mitochondrial Ca2+ uptake protein; miR, microRNA; MRAP, mitochondrial Ca2+ uniporter-regulating acidic patch; MTP, mitochondrial permeability transition; OXPHOS, oxidative phosphorylation; PAH, pulmonary arterial hypertension; PGC1α4, peroxisome proliferator-activated receptor gamma coactivator 1 alpha 4; PTP, permeability transition pore; SR, sarcoplasmic reticulum; TMH, transmembrane helix
Giorgia Pallafacchina wrote the original draft, Sofia Zanin drew the figures, and Rosario Rizzuto revised the manuscript.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)