Keywords
RNA Splice Sites,Single Nucleotide Polymorphism,Genome, Mutation,Chromosomes, Neoplasms, Information Theory, Next generation sequencing, validation
RNA Splice Sites,Single Nucleotide Polymorphism,Genome, Mutation,Chromosomes, Neoplasms, Information Theory, Next generation sequencing, validation
Next generation sequencing continues to reveal large numbers of novel variants whose impact cannot be interpreted from curated variant databases, or through reviews of peer-reviewed biomedical literature1. This has created a largely unmet need for unequivocal sources of information regarding the molecular phenotypes and potential pathology of variants of unknown significance (VUS); in cancer genomes, such sources are critically needed to assist in distinguishing driver mutations from overwhelming numbers of bystander mutations. VUS classification criteria highlight the limitations in genome interpretation due to ambiguous variant interpretation. Of the 458,899 variant submissions in NCBI’s ClinVar database with clinical interpretations, nearly half (n=221,271) are VUS (as of November 5th 2018). Only 10,784 variants in ClinVar have been documented to affect mRNA splicing at splice donor or acceptor sites, with 1,063 of these being classified as VUS, and cryptic mRNA splicing mutations are not explicitly described. The current ACMG criteria2 for variant pathogenicity prevent clinical classification of most VUS. Functional evidence that VUS either disrupt or abolish expression of genes has been sought to improve classification and provide insight into the roles, if any, of individual VUS in predisposing or causing disease. We present a comprehensive data repository for a relatively common mutation type (cis-acting variants that alter mRNA splicing). Mutations are predicted with information theory-based analyses3, and supported with functional evidence that variants in tumor genomes are specifically associated with abnormally spliced mRNAs that are infrequent or absent in transciptomes lacking these variants4.
Information theory (IT) has been proven to accurately predict impact of mutations on mRNA splicing, and has been used to interpret coding and non-coding mutations that alter mRNA splicing in both common and rare diseases3,5–15. We have described an IT-based framework for the interpretation and prioritization of non-coding variants of uncertain significance, which has been validated in multiple studies involving novel variants in patients with history or predisposition to heritable breast and/or ovarian cancer11–15.
The Cancer Genome Atlas (TCGA) Pan-Cancer Atlas (PCA) is a comprehensive integrated genomic and transcriptomic resource containing data from >10,000 tumors across 33 different tumor types16. Here, we utilized IT-based tools for assessment of high quality sequenced variants in TCGA patients for their potential impact on mRNA splicing. The accuracy of predicted mutations was evaluated with an algorithm we previously developed that compares transcripts from individuals carrying these variants with others lacking them. The results of these genome-wide analyses are presented using an online resource which can be queried through the Beacon Network17.
Controlled-access data was obtained with permission from the Data Access Committee at NIH for TCGA and from the International Cancer Genomics Consortium. Patient RNA sequencing BAM files (tumor and normal, when available) and their associated VCF files (GRCh37) were initially obtained from the CancerGenomeHub (CGhub). Files were later downloaded through Genomic Data Commons using the GDC Data Transfer Tool (version 1.3.0), as CGhub was decommissioned mid-project. Variants in VCF files which did not pass quality control (QC) were not analyzed.
We used the Shannon Pipeline software (which applies IT to rapidly perform high-throughput, in silico prediction of the impacts of variants on mRNA splicing)18 to analyze all QC-passing variants in VCFs from TCGA (>168 million variants) to evaluate their potential impact on splice site binding strength (changes in information content, Ri, measured in bits). Variants which were predicted to strengthen known natural sites or weaken cryptic splice sites were excluded from all subsequent analyses.
To validate the potential impact of Shannon Pipeline-flagged mutations, Veridical software analyzed genomic variants (including insertions and deletions) by comparing the RNA-Seq alignment in the region surrounding the variant with the corresponding interval in control transcriptomes (normal and tumor tissue of the same type) lacking the variant4,19. Veridical: a) counts abnormally spliced reads in RNA-Seq data (categorized as: cryptic site use, exon skipping, or intron inclusion [containing or adjacent to the flagged mutation]), b) applies the Yeo-Johnson transformation to these results, and c) determines the null hypothesis probability (p-value) that the transformed read count corresponds to normal splicing. In tumor types where normal controls were not available, a set of RNA-Seq datasets from 100 different normal tissues from TCGA were used (e.g. a combination of 5 tissue types: BRCA, BLCA, LUAD, KIRC, PRAD). Veridical results that were not significant for a particular variant (p-value > 0.05 for all of the splicing categories) were not further analyzed. After analysis, Veridical validated 351,423 unique mutations for their direct impact on mRNA splicing (Table 1). The Shannon pipeline-flagged and Veridical-filtered results were combined into a single large table (Dataset 120), the source data for the ValidSpliceMut SQL database and the associated Beacon application.
*The number of Veridical-flagged mutations in each The Cancer Genome Atlas (TCGA) cancer data set. Variants shared between multiple tissue types are counted for each category. Variant and RNA-Seq data were provided by The Cancer Genome Atlas Pan-Cancer Analysis Project16.
We created a publicly accessible Application Programming Interface (API) (https://beacon.cytognomix.com) that can be utilized to programmatically query variants passing filter thresholds described above (Dataset 120). It was built in accordance with the GA4GH Beacon v1.0.0 specification, which describes a Representational State Transfer (REST) API for genetic data sharing. A Beacon accepts queries using an HTTP request and returns JavaScript Object Notation (JSON). Our Beacon implementation is coded in PHP 7.0 and utilizes a MySQL database (version: 5.7.24) with indexes applied to variant ID, chromosome, and coordinate fields (GRCh37). The returned JSON object reports whether the variant was found within our Beacon dataset as well as metadata including splice site coordinate, splice type, site type, the IT-based measures Ri,initial and Ri,final, affected individual IDs, tumor type, Veridical evidence by type annotated with significance level, and, if known, the corresponding rsID with its average heterozygosity (dbSNP 150). The metadata for each variant sent to the Beacon Network is a concise subset of available results in our database. It includes the first relevant database entry, meaning that if the variant exists within multiple individuals only the first will contribute fields to the metadata. However, among this metadata is a hyperlink to our local website containing results for any remaining tumors.
We developed the website ValidSpliceMut (Figure 1) to serve as a local interface to our Beacon, allowing users to manually search for a variant, by gene name or genome coordinate range. ValidSpliceMut automatically queries our Beacon, and formats the results of the search, if any. This website provides a complete view of variants, including Veridical-based evidence on all data related to every affected individual. If a variant is associated with multiple splice sites, the user is presented with a brief overview of all affected sites and must select a desired site to continue. To obtain the coordinate of the queried variant in gene-centric notation, a link is provided which queries the Mutalyzer API and generates coordinates for all available transcripts. ValidSpliceMut only reports transcripts for the gene affected by the variant.
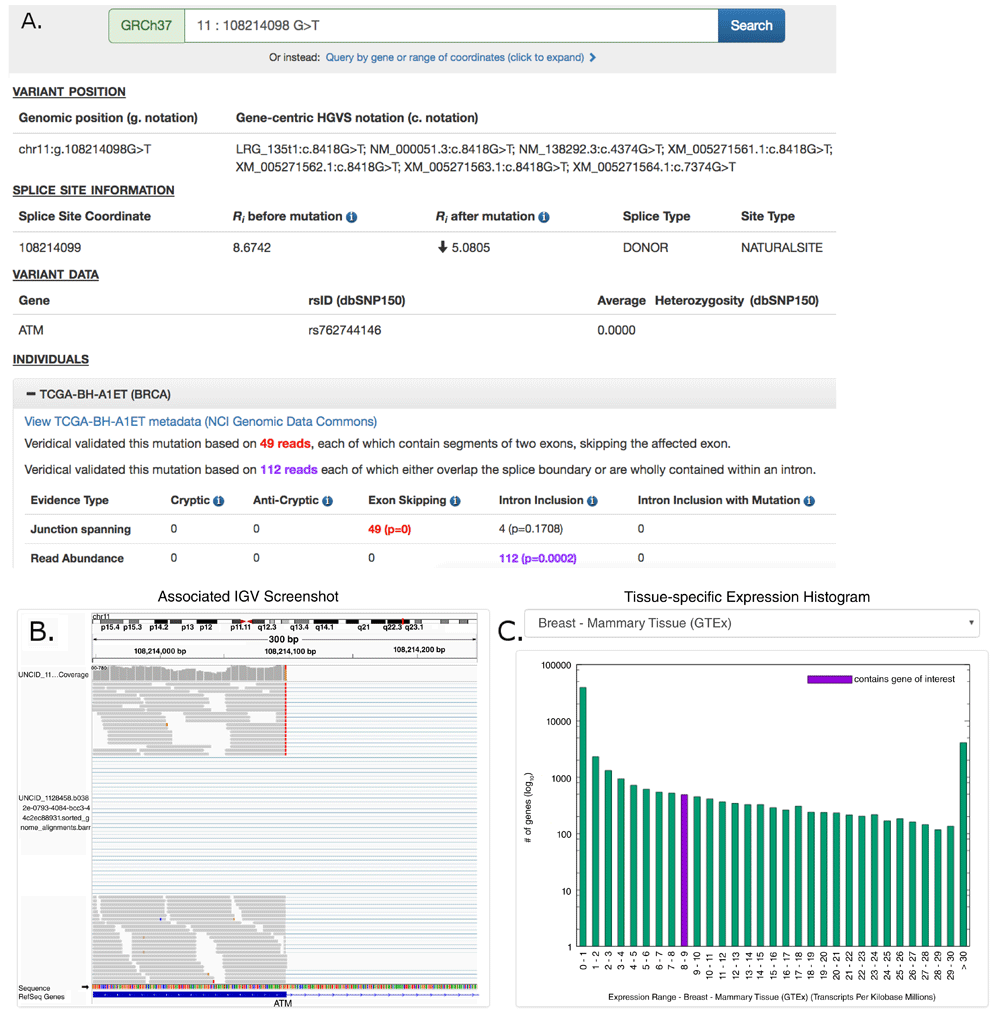
(A) The ‘Variant Position’ heading displays the variant of interest in g. notation, and provides a link which queries the Mutalyzer API to obtain the variant coordinate in a gene-centric c. mutation format. Variant-specific and splice site-specific tabular results are presented under the headings “Splice Site Information” and “Variant Data”. Results are organized by TCGA sample IDs harboring the mutation within a series of expandable panels. A link is provided to patient tumor metadata on the GDC data portal. Each panel consists of read counts and p-values by Veridical evidence type. Significant p-values (≤ 0.05) are highlighted in bold. Evidence types deemed “strongly corroborating” (Viner et al. 2014) are color coded and correspond to the dynamically generated text appearing above the table. (B) An integrative genome viewer (IGV) image showing alignment of expressed sequence reads. IGV screenshots are provided only for mutations present <1% of population (in dbSNP 150), with ≥ 5 junction-spanning reads, and are highly significant (p < 0.01) for cryptic splicing, exon skipping, and/or intron inclusion with mutation. A specific IGV screenshot for this sample captures the region surrounding the mutation. Here, several RNA-Seq reads show skipping of the affected exon. (C) A dynamically generated histogram presents expression levels of all genes for a selected normal tissue type. Genes are grouped into bins based on expression level, denoted on the x-axis. The number of genes present in each bin is shown on the y-axis (log10 scale). The histogram key indicates the expression range of the variant-containing gene. Tissue type can be changed via a drop-down list.
A results page presents variant-specific data in tabular format and an expandable list of panels describing the affected individuals. Each of these panels contains Veridical output in tabular format for the selected tumor, a link to the tumor metadata at US National Cancer Institute (by querying the GDC API to obtain a UUID which is used to construct a link to the GDC data portal), an Integrative Genome Viewer (IGV) screenshot containing the variant (IGV screenshots are available for selected variants, see below), and a histogram which presents the expression levels of the variant-containing gene compared to all other gene expression levels across a selected normal tissue type (created dynamically using gnuplot 5.0). The tissue expression data is provided by GTEx (downloaded on 10/22/18). However, several TCGA tumor types did not have a GTEx equivalent (CHOL, DLBC, MESO, READ, SARC, THYM and UVM). The GNF Expression Atlas 221 was downloaded from the UCSC Genome Browser and was used for expression data for both lymph nodes (DLBC) and the thymus (THYM). For the remaining tissues, expression data from the following studies were obtained from the Genome Expression Omnibus (GEO): GSE76297 (CHOL), GSE2549 (MESO), GSE15781 (READ) GSE44426 (SARC), and GSE44295 (UVM).
To generate IGV images presented on the webpage, a bash script was written to automatically load the RNA-Seq BAM file of a patient with a mutation of interest into IGV, set the viewing window within the region of interest (300nt window, centered on the variant), sorted to bring reads containing the variant of interest to the top of the screen (to increase chance of visualizing mutant splice form), followed by a screen capture. The generation and storage of IGV images for all patient-mutation pairs would be prohibitive due to limitations in time and server space requirements. Therefore IGV images showing evidence of splicing abnormalities were generated only for patient-mutation pairs which met the most stringent criteria: the mutation was required to be flagged for junction-spanning cryptic site use, exon skipping, or intron inclusion (with mutation); the flagged category must include 5 or more reads in this category; if the variant is present in the dbSNP database (release 150), the frequency was required to be < 1% of the population; and the Veridical results, in which the mutations flagged were required to exhibit p ≤ 0.01 for at least one form of evidence of a splicing abnormality. In some cases, the splicing event observed by Veridical may not be present within the image window as the automated procedure used to create these images does not present all evidential sequence reads due to limitations on the number of reads that are shown. Additionally, reads appearing as exon skipping may instead indicate a pre-existing cryptic site outside of the viewing window (see Table 2; FAT1:g.187521515C>A [c.11641-1G>T] and SMAD3:g.67482748C>G [c.1155-3C>G]).
| Gene | Splice Mutation | R i (bits) | Tumor | Observed Splicing Event |
|---|---|---|---|---|
| CASC5 |
15:40942786G>A (c.6212+5G>A) | 4.8 > 1.7 (Natural Site) | AML | The natural donor site of CASC5 exon 19 (NM_144508.4) is weakened, leading to a significant increase in intron inclusion. |
| DNMT3A |
2:25467022A>G
(c.1851+2T>C) | 3.6 > -3.5 (Natural Site) | AML | The natural donor site of DNMT3A exon 15 (NM_022552.4) is abolished, resulting in a significant increase in total exon skipping and intron inclusion. |
| STAG2 |
X:123176495G>A
(c.462G>A) | 6.5 > 3.5 (Natural Site) | BLCA | The natural donor of STAG2 exon 6 (NM_006603.4) is weakened, and a significant amount of exon 6 skipping is observed. |
| STAG2 |
X:123200024G>A
(c.2097-1G>A) | 19.5 > 8.6 (Natural Site) | BLCA | The natural acceptor of STAG2 exon 21 (NM_006603.4) is weakened, resulting in a significant increase in exon 21 skipping. |
| ATM |
11:108214098G>T
(c.8418G>T) | 8.7 > 5.1 (Natural Site) | BRCA | A natural donor site is weakened, leading to a significant increase in ATM exon 57 (NM_000051.3) skipping events. Some reads with mutation are involved in wildtype splicing (leaky splicing). |
| BARD1 |
2:215645882A>T
(c.716T>A) | 0.9 > 3.1 (Cryptic Site) | BRCA | The mutation strengthens a cryptic site within BARD1 exon 4 (NM_000465.2). Reads which use activated cryptic site contain the mutation (one exception). Some reads with mutation are involved in wildtype splicing (leaky splicing). |
| GATA3 |
10:8115701G>C
(c.1048-1G>C) | 0.9 > -10.7 (Natural Site) | BRCA | The mutation abolishes the natural acceptor of GATA3 exon 6 (NM_002051.2). This both increases the use of a pre-existing exonic cryptic splice site (4.2 > 5.6 bits; leads to an 8nt deletion) and significantly increases total intron inclusion. |
| TP53 |
17:7577609C>T
(c.673-1G>A) | 6.0 > -4.9 (Natural Site) | BRCA | A natural acceptor site is abolished, activating a cryptic site 49nt upstream (Ri=5.2 bits) of TP53 exon 7 (NM_000546.5). |
| POLD1 |
19:50920353A>G
(c.3119A>G) | 8.6 > 6.1 (Natural Site) | COAD | The natural donor of POLD1 exon 25 (NM_002691.3) is weakened, leading to a significant increase in total exon skipping. |
| SMAD3 |
15:67482748C>G
(c.1155-3C>G) | 11.9>3.1|-4.0 > 7.7 (Natural | Cryptic) | COAD | This mutation both weakens the natural acceptor of SMAD3 exon 9 (NM_005902.3) and creates a cryptic site (does not appear to be used). A significant amount intron inclusion reads are observed. Use of a distant pre-existing cryptic acceptor (9.6 bits; 3598nt from natural acceptor) was. |
| PIK3R1 |
5:67591246A>G
(c.936-2A>G) | 7.5 > -7.3 (Natural Site) | GBM | The natural acceptor of PIK3R1 exon 8 (NM_181504.3) is abolished, which promotes a significant increase in exon 8 skipping. |
| FAT1 |
4:187521515C>A
(c.11641-1G>T) | 5.3 > -2.4 (Natural Site) | HNSC | The natural acceptor of FAT1 exon 22 (NM_005245.3) is abolished, resulting in both intron inclusion (total intron inclusion and the use of a 2.3 bit cryptic site 82nt upstream of natural acceptor) and use of two exonic cryptic sites (237nt and 234nt from natural acceptor; Ri=1.0 bits and -0.2 bits, respectively). |
| TGFBR2 |
3:30729875G>A
(c.1397-1G>A) | 8.4 > -2.5 (Natural Site) | HNSC |
TGFBR2 exon 6 natural acceptor (NM_003242.5) is abolished, leading to multiple splicing events: intron inclusion, use of three cryptic sites (35nt exonic [Ri=3.7 bits], 30nt and 972nt intronic [Ri=0.4 bits and 11.2 bits, respectively]), and exon 6 and 7 skipping (uses a novel exon ~55kb downstream of exon 7). |
| PBRM1 |
3:52682355C>G
(c.813+5G>C) | 6.8 > 2.9 (Natural Site) | KIRC | The natural donor of PBRM1 exon 8 (NM_018313.4) is weakened, which leads to a significant increase in exon 8 skipping. |
| PBRM1 |
3:52685756A>G
(c.714+2T>C) | 7.7 > 0.7 (Natural Site) | KIRC | The natural donor of PBRM1 exon 7 (NM_018313.4) is abolished, resulting in a significant increase in total exon skipping. |
| SETD2 |
3:47079269T>A
(c.7239-2A>T) | 9.8 > 2.1| 6.4 > 9.0 (Natural | Cryptic) | KIRC | This mutation both significantly weakens the natural acceptor of SETD2 exon 18 (NM_014159.6) while strengthening a 4nt exonic cryptic site, which is used. |
| RB1 |
13:49027249T>A
(c.1814+2T>A) | 4.9 >-13.7 (Natural Site) | LUAD | The natural donor of RB1 exon 18 (NM_000321.2) is abolished, leading to a significant increase in both exon skipping and intron inclusion. All intron inclusion reads contain the mutation of interest. |
| RBM10 |
X:47006900G>T
(c.17+3G>T) | 7.8 > 4.1 (Natural Site) | LUAD | The natural donor of RBM10 exon 2 (NM_005676.4) is weakened, leading to a significant increase in exon 2 skipping. |
| RBM10 |
X:47028898G>T
(c.201+1G>T) | 8.7 > -9.9 (Natural Site) | LUAD |
RBM10 exon 3 (NM_005676.4) natural donor is abolished. RNAseq reads which overlap the exon-intron junction are observed (all reads contain mutation). Use of cryptic donor (61nt upstream of donor; Ri=1.7 bits) is observed as well. |
| DDX5 |
17:62500098 TACAG>T
(c.441+2delACAG) | -1.3 > 5.4 (Cryptic Site) | PRAD | The mutation creates a 5.4 bit cryptic donor within DDX5 exon 4 (NM_004396.3), which would lead to a 4nt deletion of exon 4. Note that wildtype splicing is still the dominant isoform observed. |
| PTEN |
10:89690802G>A
(c.210-1G>A) | 8.5 > -2.3 (Natural Site) | PRAD | The natural acceptor of PTEN exon 5 (NM_000314.4) is abolished, leading to an increased amount of total exon 5 skipping. |
| NRAS |
1:115258669A>G
(c.111+2T>C) | 8.1 > 1.1 (Natural Site) | SKCM | The mutation abolishes the natural donor of NRAS exon 2 (NM_002524.4), which promotes a significant increase in exon 2 skipping |
| PPP6C |
9:127933364C>T
(c.171G>A) | 6.7 > 3.7 (Natural Site) | SKCM | The mutation weakens PPP6C exon 2 (NM_002721.4) natural donor, leading to increased intron inclusion. All reads which cross the junction contain the mutation. A intronic cryptic site is also activated (110nt downstr.; Ri=6.4 bits). |
| PPP6C |
9:127923119C>G
(c.237+1G>C) | 6.8 > -11.8 (Natural Site) | SKCM | This mutation abolishes the natural donor of PPP6C exon 3 (NM_002721.4), resulting in a significant increase in exon 3 skipping. |
| BAP1 |
3:52442512T>C
(c.233A>G) | 1.9 > 5.1 (Cryptic Site) | UVM | A cryptic donor within BAP1 exon 4 (NM_004656.3) is strengthened, leading to a significant increase in its use. Its use leads to a 27 nt deletion of exon 4. |
Example mutations which alter splicing in tumor-associated genes found in patients with the same tumor type. Mutations are linked to their page on https://validsplicemut.cytognomix.com/, which provides additional material such as RNAseq images of the regions of interest. GRCh37 coordinates provided.
We have derived a GA4GH-standardized, searchable web resource for a large set of validated mRNA splicing variants present in diverse tumor types. All variants passing QC in TCGA cancer patients were analyzed with the Shannon pipeline18. This revealed that 1,297,242 variants were predicted to have significant impacts on normal mRNA splicing (347,549 natural and 985,112 cryptic splice sites; 35,419 affecting both types). Subsequent RNA-Seq analysis with Veridical4 provided evidence of abnormal gene expression specifically associated with a subset of these variant(s), identifying 351,423 unique mutations. Results are searchable through either the Beacon Network, or our publicly-accessible webpage.
Our results contrast with another TCGA study that investigated alternative mRNA splicing22 and demonstrated a limited set of non-constitutive exon-exon junctions attributable to cis-acting splicing mutations (n = 32). The 2,736 novel or rare variants that we report which specifically activate cryptic splicing (significant ‘junction-spanning cryptic site use’ reads found by Veridical), exceed the number reported in another study that analyzed all available TCGA tumor transcriptomes (n=1,964)23.
Validated variants (which we define as mutations) were also tallied by tumor tissue type in our study (Table 1). 33.6% of unique mutations (n=117,951) significantly weaken natural splice sites, while 69.6% (n=244,415) strengthen novel or pre-existing cryptic sites. 242,983 mutations (69%) are absent from dbSNP 150. 73,975 mutations (21%) are present in <1% of the population, of which 27,803 of these (and those not present in dbSNP) were present in multiple tumor types. Valid mutations lacking rsIDs represent either novel or recently observed variants. This low level of dbSNP saturation is consistent with the idea that many currently unknown mRNA splicing mutations may yet be discovered through additional sequencing studies.
In Table 2, we highlight a subset of validated splicing mutations (n=25) which were identified in known driver genes implicated in the COSMIC (Catalogue Of Somatic Mutations In Cancer) Cancer Gene Census catalog (CGC)24. These mutations are associated with either increased exon skipping, intron inclusion, and/or cryptic site use. Mutations in Table 2 are hyperlinked to the ValidSpliceMut webpage which provides additional information, including expression evidence supporting predictions made by the Shannon pipeline.
Many mutations generated multiple types of abnormal read evidence present in mis-spliced transcripts. Interestingly, a subset of mutations (n=28) produced evidence for every type of abnormal splicing reported by Veridical. Dataset 225 (see Data Availability) describes 11 representative mutations that simultaneously increase exon skipping, intron inclusion, and activate (or significantly increase utilization of) a strengthened cryptic site. In all but one instance, the mutation weakens the natural site while simultaneously strengthening a nearby cryptic site. The one exception involves the gene SAP30BP, where simultaneously occurring mutations in the same read (in linkage disequilibrium; separated by 4 nucleotides) independently cause two separate splicing changes: g.73702087G>A (c.661-1G>A; abolishes the natural acceptor of exon 10) and g.73702091G>A (c.664G>A; creates a weak cryptic acceptor site). The combined splicing impact of these variants is significant exon skipping, intron inclusion, and use of the activated cryptic site.
Because of the requirement for expression validation, this resource presents a set of splicing abnormalities in which we have the highest confidence. We anticipate that some correct predictions of the Shannon pipeline may have not been validated by Veridical due to the limitations of mRNA detection; for example, either low expression of the gene harboring the mutation or nonsense-mediated decay of the corresponding transcript could be consistent with the effects of a valid splicing mutation, but in the absence of a sufficient number of abnormal reads, the mutation could not be confirmed. Furthermore, at the time that the current analysis was performed, the available Shannon pipeline version did not report regulatory splicing variants adjacent to constitutive and cryptic splice sites which influence exon definition. Due to the substantial processing required for the complete TCGA dataset, the present analysis does not incorporate the effects of these variants on exon definition, which we have modeled by IT6; it does not predict the relative abundance of leaky, natural and cryptic isoforms, though such information might be inferred from the expression data on each tumor. The current version of Shannon pipeline does integrate predictions of splicing regulatory sequences and accounts for relative abundance of mRNA isoforms by exon definition, and is available through the MutationForecaster system.
The Validated Splicing Mutation resource should substantially contribute to reducing the number of outstanding VUS in tumor (and possibly some germline) genomes, and substantially increases the number of functional variants with previously unappreciated consequences to mRNA splicing, in particular, activation of cryptic splice sites. In our previous study19, a subset of the TCGA breast cancer patient data was evaluated with IT-based tools, identifying 988 mutations as significantly altering normal splicing by Veridical (19% of total mutations flagged by IT). This database greatly expands the size of the repository. Here, a higher ratio of rare or novel mutations have been validated by Veridical (24% of total mutations were flagged by IT). The higher yield found could be related to the same mutation being present in multiple samples from the same tumor type and other tumor tissues, which would be expected to increase the probability of observing abnormally expressed splice forms for the mutation.
An earlier version this article is available from bioRxiv: https://doi.org/10.1101/47445226
Archieved code and scripts used as part of this study are available from Zenodo,
Zenodo: Validated Splicing Mutations Beacon API http://doi.org/10.5281/zenodo.157989827
Zenodo: Validated Splicing Mutations Website http://doi.org/10.5281/zenodo.157982228
Zenodo: Expression Data Processing, Histogram input generation and IGV Bash Script Generating Programs http://doi.org/10.5281/zenodo.158242129
All software is licensed under a Creative Commons Attribution-Non Commercial-ShareAlike 4.0 International Public License
Zenodo: Dataset 1. Validated natural and cryptic mRNA splicing mutations. Source data computed by the Shannon pipeline and Veridical, displayed on the ValidSpliceMut website (https://validsplicemut.cytognomix.com/). DOI: http://doi.org/10.5281/zenodo.148821120
Zenodo: Dataset 2. Mutations which lead to multiple types of aberrant splicing. Representative set of mutations which significantly alter splicing in all evidence types analyzed by Veridical (i.e. cryptic splice site use, exon skipping, intron inclusion). Mutations are linked to their page on https://validsplicemut.cytognomix.com/, which provides additional material such as RNA-Seq images of the regions of interest. DOI: https://dx.doi.org/10.5281/zenodo.148994125
License: CC0 1.0
Controlled-access TCGA sequence data was accessed with permission from NCBI (dbGaP Project #988: “Predicting common genetic variants that alter the splicing of human gene transcripts”; Approval Number #13930-11; PI: PK Rogan) and the International Cancer Genome Consortium (ICGC Project #DACO-1056047; “Validation of mutations that alter gene expression”).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)