Keywords
genomics, transcriptomics, proteomics, RNA-seq, microarray, database, identifiers
This article is included in the RPackage gateway.
genomics, transcriptomics, proteomics, RNA-seq, microarray, database, identifiers
Since the advent of genome sequencing projects, many technologies have been developed to get access to different molecular information on a large scale and with high throughput. DNA micro-arrays are probably the archetype of such technology because of their historical impact on gathering data related to nucleic acids: genomic DNA and RNA. They triggered the emergence of “omics” fields of research such as genomics, epigenomics or transcriptomics. Lately massive parallel sequencing further increased the throughput of data generation related to nucleic acids by several orders of magnitude. In a different way, mass spectrometry-related technologies allow the identification and the quantification of many kinds of molecular entities such as metabolites and proteins. Many information systems have been developed to manage the exploding amount of data and knowledge related to biological molecular entities. These resources manage different aspects of the data. For example some are genome or proteome centered, whereas others are focused on molecular interactions and pathways. Thus all these resources rely on different identifier systems to organize the concepts of interest. The value of all the experimental data and all the knowledge collected in public or private resources is very high as such but is also often synergistically leveraged by their cross comparison in a dedicated manner. Indeed many datasets can be relevant when addressing the understanding of a specific biological system, a phenotypic trait or a disease for example. These datasets can focus on different biological entities such as transcripts or proteins in different tissues, conditions or organisms. Comparing all these data and integrating them with available knowledge requires the ability to map the identifiers on which each resource relies.
To achieve this task public and proprietary information systems provide mapping tables between their own identifiers and those from other resources. Furthermore many tools have been developed to facilitate the access to this information. Ensembl BioMarts (Kinsella et al., 2011), mygene (Wu et al., 2013), and g:Profiler (Reimand et al., 2016a) are popular examples among many others. However, as pointed out by van Iersel et al. (2010), these tools are generally dedicated to a particular domain and not necessarily relevant or complete for all research projects, and keeping them up-to-date can also be an issue. Recognizing these challenges van Iersel et al. (2010) proposed the BridgeDb framework providing to bioinformatics developers a standard interface between tools and mapping services and also allowing the easy integration of custom data by a transitivity mechanism.
Here we present BED: a biological entity dictionary. BED has been developed to address three main challenges. The first one is related to the completeness of identifier mappings. Indeed direct mapping information provided by the different systems are not always complete and can be enriched by mappings provided by other resources. More interestingly direct mappings not identified by any of these resources can be indirectly inferred by using mappings to a third reference. For example, many human Ensembl gene identifiers are not directly mapped to any Entrez gene identifiers but such mapping can be inferred using respective mappings to HGNC identifiers. The second challenge is related to the mapping of deprecated identifiers. Indeed entity identifiers can change from one resource release to another. The identifier history is provided by some resources, such as Ensembl or the NCBI, but it is generally not used by mapping tools. The third challenge is related to the automation of the mapping process according to the relationships between the biological entities of interest. Indeed mapping between gene and protein identifier scopes should not be done the same way than two scopes of gene identifiers. Also converting identifiers from different organism should be possible using gene ortholog information.
To meet these challenges we designed a graph data model describing possible relationships between different biological entities and their identifiers. This data model has been implemented with the Neo4j® graph database (Neo4j inc, 2017) and conversion rules have been defined and coded in an R (R Core Team, 2017) package. We provide an instance of the BED database focused on human, mouse and rat organism but many functions are available to construct other instances tailored to other needs.
The BED (Biological Entity Dictionary) system relies on a data model inspired by the central dogma of molecular biology (Crick, 1970) and describing relationships between molecular concepts usually manipulated in the frame of genomics studies (Figure 1). A biological entity identifier (BEID) can identify either a Gene (GeneID), a Transcript (TranscriptID), a Peptide (PeptideID) or an Object (ObjectID). Object entities can correspond to complex concepts coded by any number of genes (i.e. a protein complex or a molecular function). BEID are extracted from public or private databases (BEDB). BEDB can provide an Attribute related to each BEID. For example it can be the sequencing region provided by the Ensembl database (Zerbino et al., 2018) or the identifier status provided by Uniprot (The UniProt Consortium, 2017). BEID can have one or several associated names (BENames) and symbols (BESymbol). GeneID can have one or several homologs in other organisms belonging to the same GeneIDFamily. Many genomics platforms, such as micro-array, allow the identification of biological entity by using probes identified by ProbeID. In general, BEID can be targeted by several probes belonging to a Platform which is focused on one, and only one, type of entity (BEType) among those described above: Gene, Transcript, Peptide or Object. A BEType can have several BEType products but can be the product of at most one BEType. This constraint allows the unambiguous identification of the most relevant path to convert identifiers from one scope to another and is fulfilled by the current data model: peptides are only produced from transcripts, which are only produced from genes, which can also code for objects.
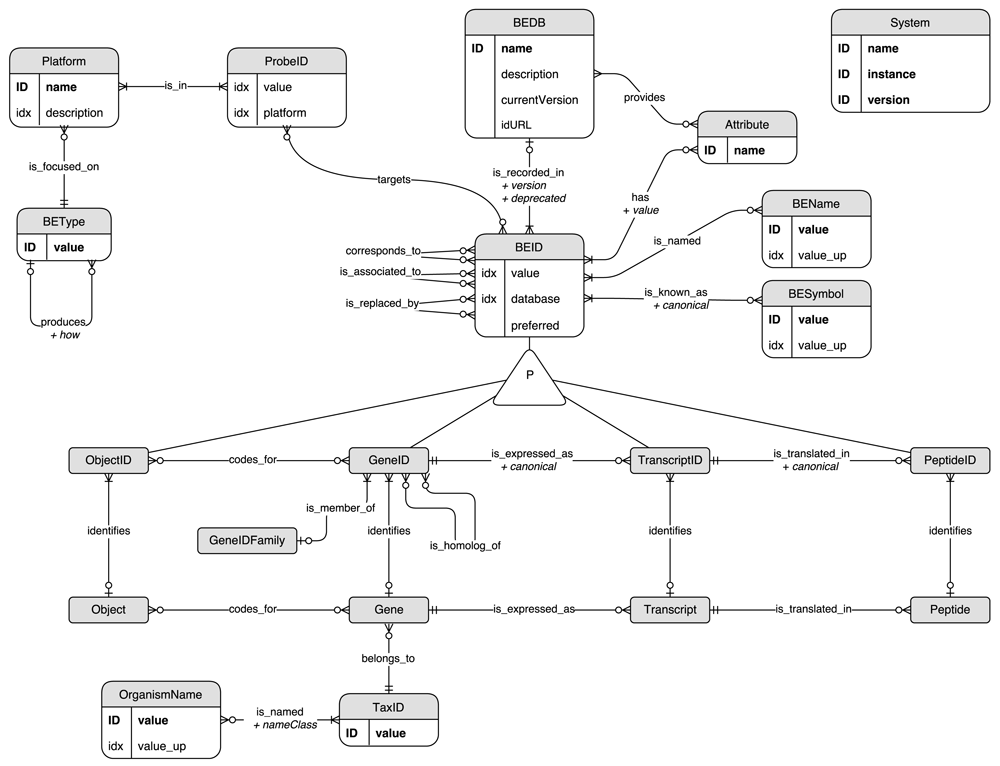
The model is shown as an Entity/Relationship (ER) diagram: entities correspond to graph nodes and relationships to graph edges. “ID” and “idx” indicate if the corresponding entity property is unique or indexed respectively. Some redundancies occur in this data model. Indeed some “value” properties are duplicated in upper case (“value_up”) in order to improve the performance of case-insensitive searches. Also the database of a BEID node is provided as a property to ensure uniqueness of the couples of “database” and “value” properties. The same approach has been applied for the “platform” property of ProbeID nodes.
BEID identifying the same biological entity are related through three different kinds of relationship according to the information available in the source databases, and to the decision made by the database administrator about how to use them. Two BEID which corresponds_to each other both identify the same biological entity. A BEID which is_associated_to or which is_replaced_by another BEID does not directly identify any biological entity: the link is always indirect through one or several other BEID. Therefore, by design a BEID which is_associated_to or which is_replaced_by another BEID can be related to several different biological entities. It is not the case for other BEID which identify one and only one biological entity. This set of possible relationship allows the indirect mapping of different identifiers not necessarily provided by any integrated resource.
In order to efficiently leverage an indirect path through these different relationships the data model has been implemented in a Neo4j® graph database (Neo4j inc, 2017).
Two R (R Core Team, 2017) packages have been developed to feed and query the database. The first one, neo2R, provides low level functions to interact with Neo4j®. The second R package, BED, provides functions to feed and query the BED Neo4j® graph database according to the data model described above.
Many functions are provided within the package to build a tailored BED database instance. These functions are not exported in order not to mislead the user when querying the database (which is the expected most frequent usage of the system). An R markdown document showing how to build a BED database instance for human, mouse and rat organisms is provided within the package. It can be adapted to other organisms or needs.
Briefly these functions can be divided according to three main levels:
The lowest level function is the bedImport function which loads a table in the Neo4j® database according to a Cypher® query.
Functions of the second level allow loading identifiers and relationships tables ensuring the integrity of the data model.
Highest level functions are helpers for loading information provided by some public resources in different specific formats.
The BED R package provides several functions to retrieve identifiers from different resources, and also to convert identifiers from one reference to another. These functions generate and call Cypher® queries on the Neo4j® database. Converting thousands of identifiers can take some time (generally a few seconds). Also such conversions are often recurrent and redundant. In order to improve the performance for such recurrent and redundant queries, a cache system has been implemented. The first time, the query is run on Neo4j® for all the relevant ID related to user input and the result is saved in a local file. Next time similar queries are requested, the system does not call Neo4j® but loads the cached results and filters it according to user input. By default the cache is flushed when the system detects inconsistencies with the BED database. It can also be manually flushed if needed.
Minimal system requirements for running BED and neo2R R packages:
The graph database has been implemented with Neo4j® version 3 (Neo4j inc, 2017). The BED R package depends on the following packages available in the Comprehensive R Archive Network (CRAN):
visNetwork (Almende et al., 2017)
dplyr (Wickham et al., 2017)
htmltools (RStudio inc, 2017)
DT (Xie, 2016)
shiny (Chang et al., 2017)
miniUI (Cheng, 2016)
rstudioapi (Allaire et al., 2017)
An instance of the BED database (UCB-Human) has been built using the script provided in the BED R package and made available in a Docker® image (Docker inc, 2017) available here: https://hub.docker.com/r/patzaw/bed-ucb-human/
This instance used to exemplify the following use cases is focused on Homo sapiens, Mus musculus and Rattus norvegicus organisms and it has been built from the following resources:
Ensembl (Zerbino et al., 2018)
Uniprot (The UniProt Consortium, 2017)
biomaRt (Durinck et al., 2009)
GEOquery (Davis & Meltzer, 2007)
Clarivate Analytics MetaBase® (Clarivate Analytics, 2017)
The numbers of biological entity (BE) identifiers (BEID) available in this BED database instance and which can be mapped to each other are shown in Table 1. In total, 3,519,181 BEID are available in this BED instance. This number includes deprecated identifiers without successor and which therefore cannot be mapped to any other identifier. All the genomics platforms included in this BED database instance are shown in Table 2. They provide mapping to BEID from 354,205 ProbeID in total.
Numbers have been split according to the BE type and the organism. Only BEID which can be mapped to each other are taken into account (e.g. excluding deprecated identifiers without successor).
| BE | Organism | Database | BEID | URL |
|---|---|---|---|---|
| Gene | Homo sapiens | MIM_GENE | 17,146 | http://www.omim.org |
| Gene | Homo sapiens | miRBase | 1,881 | http://www.mirbase.org |
| Gene | Homo sapiens | UniGene | 23,012 | https://www.ncbi.nlm.nih.gov |
| Gene | Homo sapiens | Ens_gene | 68,460 | http://www.ensembl.org |
| Gene | Homo sapiens | HGNC | 41,195 | http://www.genenames.org |
| Gene | Homo sapiens | EntrezGene | 81,761 | https://www.ncbi.nlm.nih.gov |
| Gene | Homo sapiens | Vega_gene | 19,141 | http://vega.sanger.ac.uk |
| Gene | Homo sapiens | MetaBase_gene | 23,377 | https://portal.genego.com |
| Gene | Mus musculus | miRBase | 1,193 | http://www.mirbase.org |
| Gene | Mus musculus | UniGene | 21,576 | https://www.ncbi.nlm.nih.gov |
| Gene | Mus musculus | Ens_gene | 56,954 | http://www.ensembl.org |
| Gene | Mus musculus | MGI | 78,547 | http://www.informatics.jax.org |
| Gene | Mus musculus | EntrezGene | 103,555 | https://www.ncbi.nlm.nih.gov |
| Gene | Mus musculus | Vega_gene | 45,237 | http://vega.sanger.ac.uk |
| Gene | Mus musculus | MetaBase_gene | 20,628 | https://portal.genego.com |
| Gene | Rattus norvegicus | miRBase | 495 | http://www.mirbase.org |
| Gene | Rattus norvegicus | UniGene | 12,613 | https://www.ncbi.nlm.nih.gov |
| Gene | Rattus norvegicus | Ens_gene | 34,963 | http://www.ensembl.org |
| Gene | Rattus norvegicus | RGD | 46,976 | https://rgd.mcw.edu |
| Gene | Rattus norvegicus | EntrezGene | 57,026 | https://www.ncbi.nlm.nih.gov |
| Gene | Rattus norvegicus | Vega_gene | 1,146 | http://vega.sanger.ac.uk |
| Gene | Rattus norvegicus | MetaBase_gene | 17,505 | https://portal.genego.com |
| Transcript | Homo sapiens | Ens_transcript | 228,389 | http://www.ensembl.org |
| Transcript | Homo sapiens | Vega_transcript | 37,017 | http://vega.sanger.ac.uk |
| Transcript | Homo sapiens | RefSeq | 189,384 | https://www.ncbi.nlm.nih.gov |
| Transcript | Mus musculus | Ens_transcript | 136,967 | http://www.ensembl.org |
| Transcript | Mus musculus | Vega_transcript | 120,271 | http://vega.sanger.ac.uk |
| Transcript | Mus musculus | RefSeq | 112,390 | https://www.ncbi.nlm.nih.gov |
| Transcript | Rattus norvegicus | Ens_transcript | 42,393 | http://www.ensembl.org |
| Transcript | Rattus norvegicus | Vega_transcript | 1,271 | http://vega.sanger.ac.uk |
| Transcript | Rattus norvegicus | RefSeq | 98,431 | https://www.ncbi.nlm.nih.gov |
| Peptide | Homo sapiens | Ens_translation | 109,643 | http://www.ensembl.org |
| Peptide | Homo sapiens | Vega_translation | 36,460 | http://vega.sanger.ac.uk |
| Peptide | Homo sapiens | RefSeq_peptide | 117,465 | https://www.ncbi.nlm.nih.gov |
| Peptide | Homo sapiens | Uniprot | 232,130 | http://www.uniprot.org |
| Peptide | Mus musculus | Ens_translation | 65,406 | http://www.ensembl.org |
| Peptide | Mus musculus | Vega_translation | 57,318 | http://vega.sanger.ac.uk |
| Peptide | Mus musculus | RefSeq_peptide | 79,418 | https://www.ncbi.nlm.nih.gov |
| Peptide | Mus musculus | Uniprot | 114,825 | http://www.uniprot.org |
| Peptide | Rattus norvegicus | Ens_translation | 30,245 | http://www.ensembl.org |
| Peptide | Rattus norvegicus | Vega_translation | 1,260 | http://vega.sanger.ac.uk |
| Peptide | Rattus norvegicus | RefSeq_peptide | 68,716 | https://www.ncbi.nlm.nih.gov |
| Peptide | Rattus norvegicus | Uniprot | 40,786 | http://www.uniprot.org |
| Object | Homo sapiens | MetaBase_object | 24,748 | https://portal.genego.com |
| Object | Homo sapiens | GO_function | 4,104 | http://amigo.geneontology.org |
| Object | Mus musculus | MetaBase_object | 22,000 | https://portal.genego.com |
| Object | Mus musculus | GO_function | 4,081 | http://amigo.geneontology.org |
| Object | Rattus norvegicus | MetaBase_object | 18,648 | https://portal.genego.com |
| Object | Rattus norvegicus | GO_function | 4,001 | http://amigo.geneontology.org |
The getBeIds function returns all BE identifiers from a specific scope. A scope is defined by the type of BE or probe, the source of the identifiers (database or platform) and the organism. For example, the following code returns all the Ensembl identifiers of human genes.
beids <- getBeIds( be="Gene", source="Ens_gene", organism="human", restricted=FALSE ) head(beids)
## id preferred Gene db.version db.deprecated
## 82643 ENSG00000283891 TRUE 64781 91 FALSE
## 82642 ENSG00000207766 TRUE 64783 91 FALSE
## 82645 ENSG00000276678 TRUE 64785 91 FALSE
## 82644 ENSG00000265993 TRUE 64787 91 FALSE
## 82647 ENSG00000283793 TRUE 64789 91 FALSE
## 82646 ENSG00000283621 TRUE 64791 91 FALSEThe id column corresponds to the BEID from the source of interest. The column named according to the BE type (in this case Gene) corresponds to the internal identifiers of the related BE. This internal identifier is not a stable reference that can be used as such. Nevertheless, it is useful to identify BEID identifying the same BE. In the example above even if most of Gene BE are identified by only one Ensembl gene BEID, many of them are identified by two or more (5,809 / 59,515 = 10%); 277 BE are even identified by more than 10 Ensembl BEID (Figure 2.a). In this case, most of these redundancies come from deprecated ID from former versions of the Ensembl database (version in use here: 91) and can be excluded by setting the restricted parameter to TRUE when calling the getBeIds function (Figure 2.b). However many BE are still identified by two or more current Ensembl BEID (2,715 / 59,515 = 5%). This result comes from the way the BED database is constructed: When two identifiers from the same resource correspond to the same identifier in another resource (correspond_to relationship in the data model), all these BEID are considered to identify the same BE.
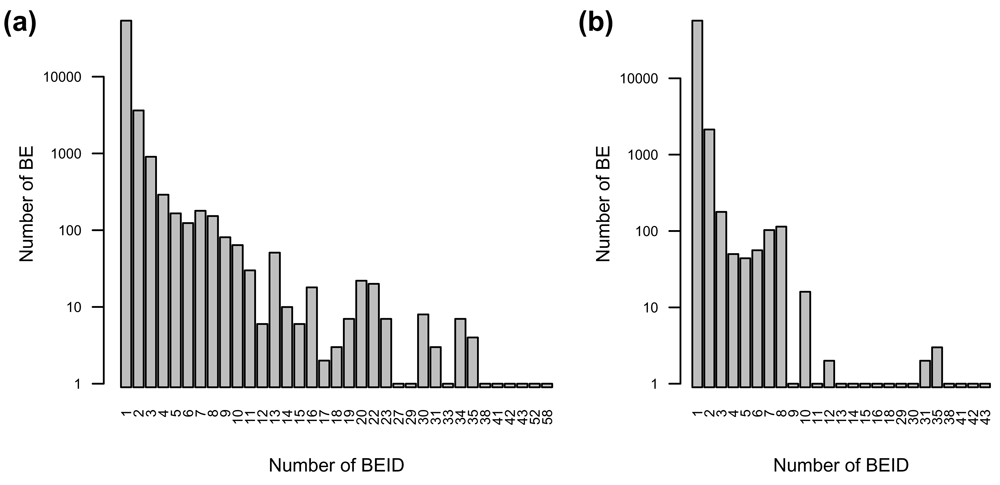
a) All Ensembl gene ID. b) Current Ensembl gene ID (version 91).
A complex example of such mapping is shown in Figure 3 mapping all the BEID of the human TAS2R8 gene which codes for a protein of the family of candidate taste receptors. There are three identifiers corresponding to this gene symbol in Ensembl. All these three identifiers correspond to the same Entrez gene and the same HGNC identifiers. All these BEID are thus considered to identify the same gene. It turns out that the three Ensembl BEID correspond to the same gene mapped on different sequence version of the chromosome 12: the canonical (ENSG00000121314), CHR_HSCHR12_2_CTG2 (ENSG00000272712) and CHR_HSCHR12_3_CTG2 (ENSG00000277316). This information provided by Ensembl is encoded in the seq_region attribute for each Ensembl BEID (see data model) and is used to define preferred BEID which are mapped on canonical version of chromosome sequences. The ENSG00000272712 identifier shows also a complex history in former Ensembl versions.
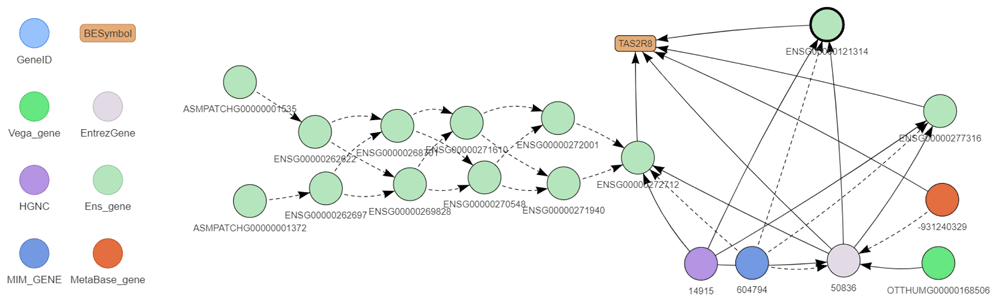
BEID are shown as circle and gene symbol in the rounded box. The color legend is shown to the left of the figure. BEID surrounded in bold correspond to preferred identifiers. Solid arrows represent correspond_to and is_known_as relationships. Dotted arrows represent is_replaced_by and is_associated_to relationships. This graph has been drawn with the exploreBe function.
The main goal of BED is to convert identifiers from one scope to another easily, rapidly and with high completeness. It has been thought in order to allow recurring comparisons to each other of many lists of biological entities from various origins.
The function guessIdOrigin can be used to guess the scope of any list of identifiers. A simple example regarding the conversion of human Ensembl gene to human Entrez gene identifiers is shown below and discussed hereafter. By setting the restricted parameter to TRUE the converted BEID are restricted to current - non-deprecated - version of Entrez gene identifiers. Nevertheless all the input BEID are taken into account, current and deprecated ones.
bedConv <- convBeIds( ids=beids$id, from="Gene", from.source="Ens_gene", from.org="human", to.source="EntrezGene", restricted=TRUE )
Among all the 68,460 human Ensembl gene identifiers available in the database, 21,718 (32%) were not converted to any human Entrez gene identifier: 21,073 (33%) of the 64,661 non-deprecated and 645 (17%) of the 3,799 deprecated identifiers.
Three other tools were used on January 04, 2018 to perform the same conversion task: biomaRt (Durinck et al., 2009; Kinsella et al., 2011), mygene (Mark et al., 2014; Wu et al., 2013), and gProfileR (Reimand et al., 2016a; Reimand, 2016b). At that time, biomaRt and mygene were based on the Ensembl 91 release whereas gProfileR was based on release 90.
The numbers of human Ensembl gene identifiers successfully converted by each method are compared in Figure 4. Five identifiers were only converted by gProfileR. They were provided by former versions of Ensembl or NCBI but are now deprecated in the current releases of these two resources. All the other gene identifiers converted by the different methods were also converted by BED. However, BED was able to map at least 17,912 more identifiers than all the other tools (Figure 4.a). A few of these mappings (3,154) are explained by the fact that BED is the only tool mapping deprecated identifiers to current versions. Nevertheless, even when focusing on the mapping of current versions of Ensembl identifiers BED was able to map 14,758 more identifiers than all the other tools (Figure 4.b). A few of these mappings (627) are directly provided by the NCBI. But most of them (14,131) are inferred from a mapping of the Ensembl and Entrez gene identifiers to the same HGNC (Gray et al., 2015) identifier.
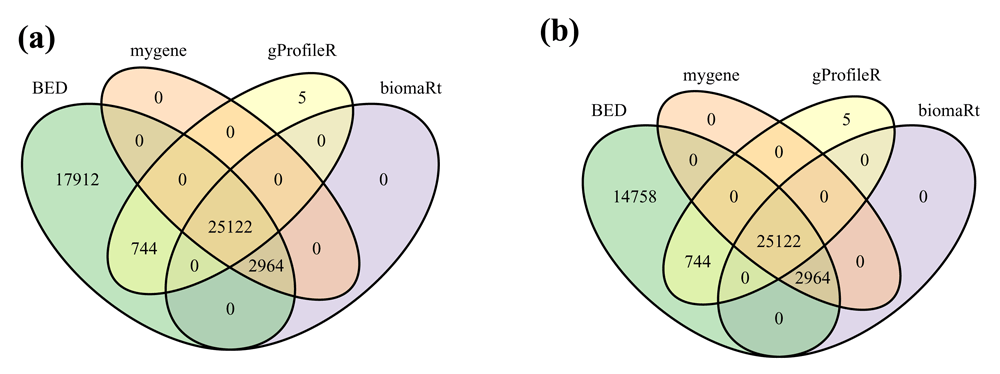
Venn diagrams showing the number of human Ensembl gene identifiers mapped to at least one human Entrez gene identifier by the different tested tools when focusing (a) on all 68,460 or (b) on current 64,661 BEID (Ensembl 91 release).
A rough approximation of running times of the different methods is provided in Table 3. The aim of this table is to show that BED, as a dedicated and locally available tool, is a very efficient option to convert large lists of identifiers on the fly and recurrently. The aim of BED is to improve the efficiency of identifier conversion in a well defined context (organism, information resources of interest. . .) and not to replace biomaRt, mygene, gProfileR or other tools which provide many more features for many organisms and which should not be narrowed to this task for a complete comparison.
| Method | Running time |
|---|---|
| BED (Not cached) | ~9.9 secs |
| BED (Cached) | ~2.5 secs |
| biomaRt | ~40 secs |
| mygene | ~3.9 mins |
| gProfileR | ~1.2 mins |
The BED convBeIds function can be used to convert identifiers from any available scope to any other one. It automatically find the most relevant path according to the considered biological entities. It allows elaborate mapping such as the conversion between probe identifiers from a platform focused on mouse transcripts into human protein identifiers. Because such mappings can be intricate, BED also provides a function to show the shortest relevant path between two different identifiers (Figure 5).
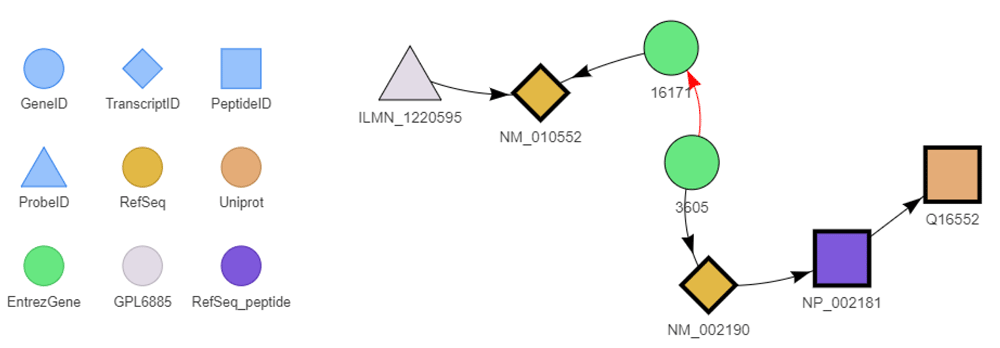
The legend is shown to the left of the figure. The red arrow represents the is_homolog_of relationship. This graph has been drawn with the exploreConvPath function.
Some additional use cases and examples are provided in the BED R package vignette. Several functions are available for annotating BEID with symbols and names, again taking advantage of information related to connected identifiers. Other functions are also provided to seek relevant identifiers of a specific biological entity. These functions are used by a shiny (Chang et al., 2017) gadget (Figure 6) providing an interactive dictionary of BEID which is also made available as an Rstudio add-in (Allaire et al., 2017; Cheng, 2016).
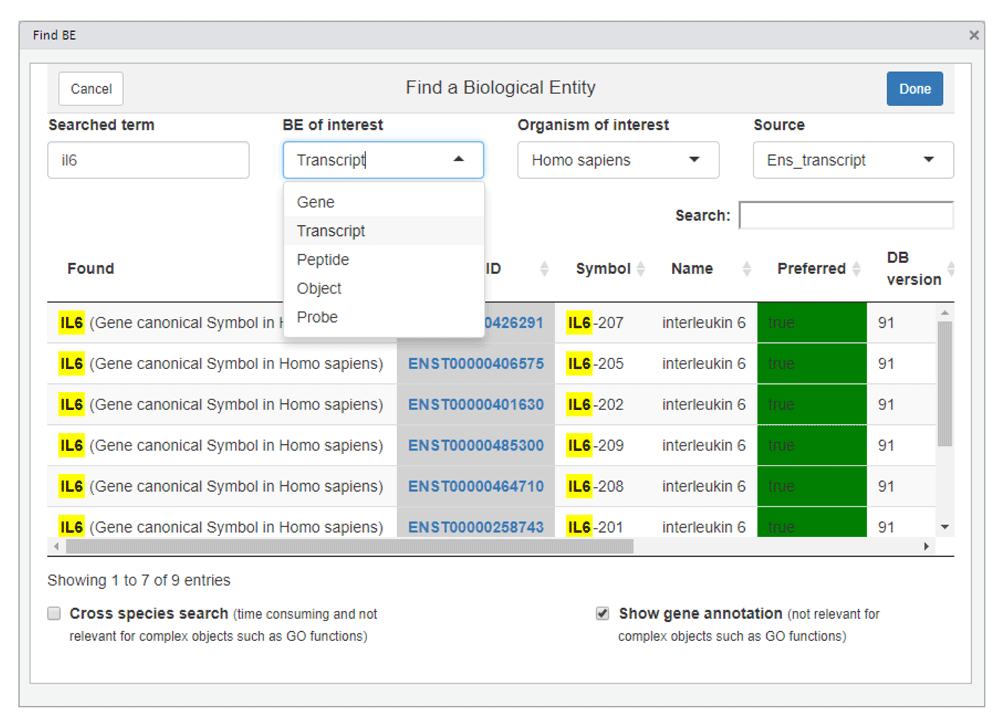
In this example the user is looking after human Ensembl transcript identifiers corresponding to “il6”.
BED is a system dedicated to the mapping between identifiers of molecular biological entities. It relies on a graph data model implemented with Neo4j® and on rules coded in an R package. BED leverages mapping information provided by different resources in order to increase the mapping efficiency between each of them. It also allows the mapping of deprecated identifiers. Rules are used to automatically convert identifiers from one scope to another using the most appropriate path.
The intent of BED is to be tailored to specific needs, and beside functions for querying the system, the BED R package provides functions to build custom instances of the database. Database instances can be locally installed or shared across a community. This design combined with a cache system makes BED efficient for converting large lists of identifiers from and to a large variety of scopes.
Because of our research field we provide an instance focused on human, mouse and rat organisms. This database instance can be directly used in relevant projects but it can also be enriched depending on user or community needs.
Latest source code is available at:
https://github.com/patzaw/neo2R
Archived source code as at time of publication:
https://zenodo.org/badge/latestdoi/119707445 (Godard, 2018a)
https://zenodo.org/badge/latestdoi/119698430 (Godard, 2018b)
Software is available to use under a GPL-3 license
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)