Keywords
Sample profile, Gene set profile, Genomics, Integrated analysis
Sample profile, Gene set profile, Genomics, Integrated analysis
This version includes revisions and response to referee report 16 Apr 2018. Mainly, we have changed the default parameter settings (under ‘Sample Profile’ and ‘Changepoint Input’ panel) on the online version of the shinySISPA web-tool to output results as shown in the manuscript. The example datasets are available for one-, two-, and three-feature analysis under ‘Data Input’ panel. We have updated Figure 1 and Figure legend pertaining to the reviewer comment. We have corrected the typo in the main text pertaining to the number of patients with and without profile activity
See the authors' detailed response to the review by Yun Zhang
Unlike gene set profiling, sample profiling is a challenge due to the heterogeneity between, and within the tumor patient samples. Identification of homogenous groups of samples or molecular subtypes is commonly approached using clustering methods (e.g., Handl et al., 2005; Kowalski et al., 2016; Monti et al., 2003; Șenbabaoğlu et al., 2014; Verhaak et al., 2010). Whether or not the sample groups are meaningful and the clustering stable, requires additional testing, is highly subjective, limited to examining changes in a single data type, and often require removal of genes or samples to obtain the desired results. While clustering tools such as TNBC subtyping (Chen et al., 2012; Lehmann et al., 2011) are convenient for subtype discovery and sample classification, they are restricted to studying a specific cancer tissue and data type, within the context of established expression signature profiles. Although these methods may prove useful in certain case, there is a need for a basic tool that can identify sample groups using any combination of genomic data types based on a gene or gene set and molecular profile of interest. Some examples of gene sets may be derived from a specific biological process, network, gene enrichment analysis, a gene panel, etc. A molecular profile is a series of increasing or decreasing changes among diverse data types operating on a given gene set. For example, a gene mutation with expression is a molecular profile of increased variant support with increased levels of expression. The shinySISPA is a web tool developed to implement the novel method, SISPA (Kowalski et al., 2016), for defining samples with similar molecular profiles based on a user input gene set and data types. SISPA does not impose analytical distribution assumptions on the data, and is scalable to define samples that support a general profile defined by any combination of genomic data types applied to any number of genes.
SISPA is written in the R programming language (R project) and the shiny web application framework is implemented using the Shiny R package (Chang et al., 2016).
The tool is hosted on a 64bit CentOS 6 server (http://shinygispa.winship.emory.edu/shinySISPA/) running the Shiny Server program designed to host R Shiny applications. This tool has been extensively tested on Windows 7 and Mac Pro 10 operating system with firefox and google chrome browser. Given a dataset of 377 samples and 16 genes under the two-feature analysis, it took three seconds to obtain shinySISPA defined sample groups and less than a second to generate the waterfall plot. The time it takes to generate sample profile diagnostic plots depends on the number of genes in a set; it took less than 10 seconds for 16 genes in both sets of a two-feature analysis. As a note, speed at which results are generated is also dependent on the internet connectivity.
The tool workflow consists of four basic inputs as shown in Figure 1:
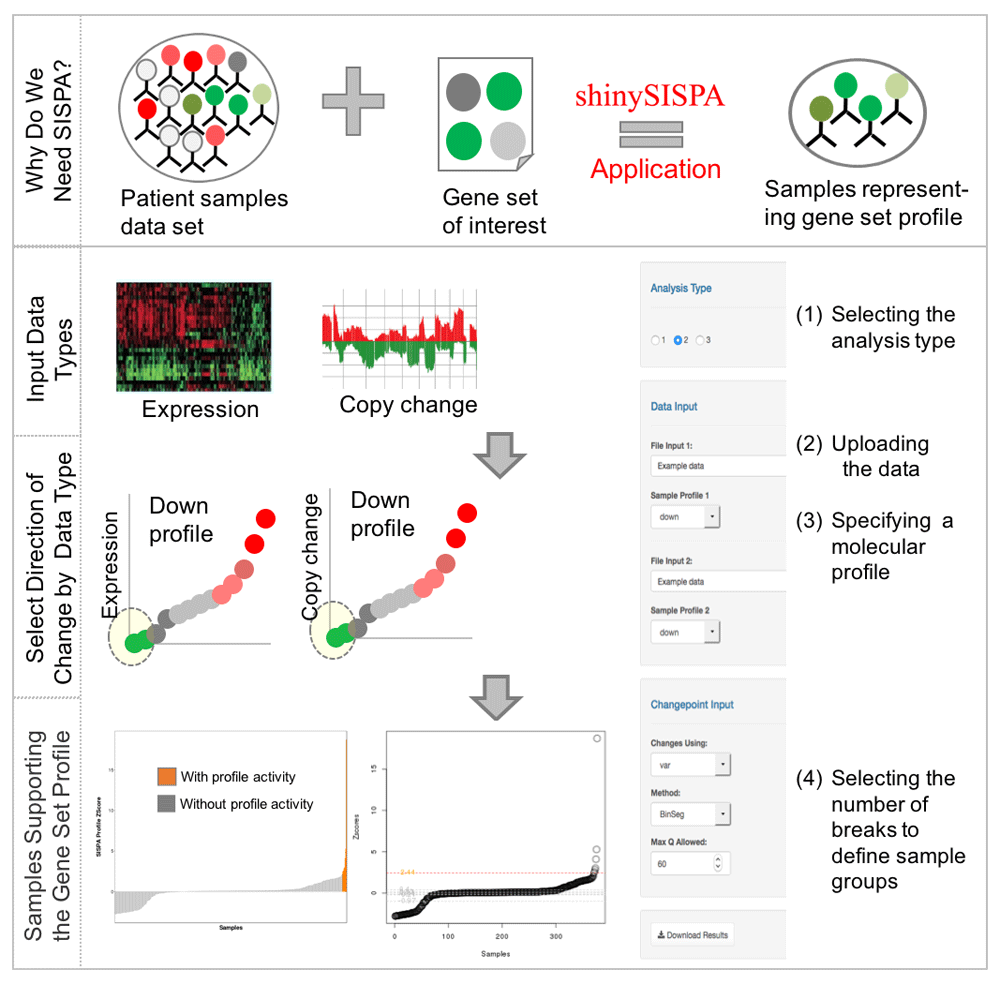
Here, we define samples supporting the molecular profile of decreased gene expression and copy loss. The tool requires user selection of analysis type, user upload of data types on samples and gene sets, and specification of a profile to output the samples supporting that profile. The samples are selected based on a change point model applied to composite (among features and genes), within-sample z-scores. A waterfall plot of profile activity is output with samples selected in orange as showing the most support for the profile.
(1) Selecting the analysis type. User selects a single-, two-, or three-feature analysis, where a feature corresponds to a specific data type (e.g., expression, methylation, mutation, copy number variation) and thus, a single-feature analysis refers to use of a single data type, while a two-feature uses a combination of two data types and so forth.
(2) Uploading the data. User inputs the data for each feature containing the genes and samples of interest. The same samples are required for each feature, though the gene sets may differ between features.
(3) Specifying a molecular profile. A molecular profile is a series of increasing (“up”) or decreasing (“down”) genomic changes within each feature. In Figure 1, a profile of decreased expression with decreased copy number is input.
(4) Selecting the number of breaks to define sample groups. User can specify the change point detection method (Killick et al., 2016) for finding optimal break points in the distribution of computed composite (among features) z-scores within samples (Kowalski et al., 2016; see Supplementary File 1).
The results are output in four separate tabs:
(1) Input Data. Summarizes the user input data in terms of the input number of genes, number of samples, and box plot distribution by data type.
(2) SISPA Results. Outputs the table of defined sample groups with their gene set enrichment score for the selected analysis type and molecular profile of interest. The scatter plot on the right displays all the change points detected within the data-set, samples falling in the topmost change point are the samples with the profile activity. The frequency plot at the rightmost bottom represents the distribution of the number of samples with and without the profile activity.
(3) Waterfall Plot. To visualize the sample groups that correlate with the profile of interest. Samples with the profile activity have the highest score and are shown in orange filled bars, while samples without the profile activity are shown with grey-filled bars.
(4) Sample Profile. Represents the diagnostic plots to visualize the distribution of the user-input data overall by the identified sample groups. It also allows the users to view data distribution for a selected gene in the set within each data type to assess what genes in particular satisfy the profile versus samples without profile.
All results generated during the process are directly downloadable on the user’s local computer. A detailed manual with tool settings are provided in the Supplementary File 1. Upon forming such sample groups, one may readily examine the effect of a profile on various clinical and biological clinical outcomes.
We applied shinySISPA to profile newly diagnosed multiple myeloma (MM) patients for decreased gene expression and copy number based on a GISPA (Gene Integrated Set Profile Analysis)-derived gene set characterizing the IgH translocation in the MM cell lines (Kowalski et al., 2016). The t(14;16) translocation is known to be associated with poor prognosis in MM. By applying the shinySISPA tool to the t(14;16) characterized gene set profile, we were able to translate cell line profiles to patient profiles. Using the IA6 release of Multiple Myeloma Research Foundation (MMRF) CoMMpass study, we downloaded data from 377 newly diagnosed patients at pre-treatment with available clinical outcomes, RNA-Seq expression, and DNA-copy number variations from the MMRF Research Gateway portal. Based on our two-feature analysis, 7 of the 370 MM patients were defined with profile activity (Figure 1) by identifying changes in variance using change point v2.2.2. Furthermore, we used CASAS (Rupji et al., 2017) to compare survival curves of the identified two sample groups for downstream clinical interpretation. We found seven samples with profile activity to be significantly (P<0.0001) associated with poor survival as compared to the 370 samples without the profile activity (HR = 9.81; 95% CI = (3.39, 28.37)).
We have demonstrated the utility of our shinySISPA tool in translating cell line characterized gene sets molecular profile to patient profiling (Kowalski et al., 2016); however, one can use any a priori-defined gene sets with any combination of molecular data for identifying samples with a similar gene set profile. The introduction of a change point model to select samples with profile support offers a more objective approach than with clustering methods. With only a gene set and a combination of data types from the same samples, the tool is widely applicable to many settings. For example, shinySISPA may be used to define patients based on known drug targets and pathways, or to identify patients that may be at risk for poor prognosis based on known prognostic markers.
The example sample data used to demonstrate shinySISPA workflow is available on the web-version and with the package source code at: https://github.com/BhaktiDwivedi/shinySISPA.
The shinySISPA software is available at http://shinygispa.winship.emory.edu/shinySISPA/
The stand-alone version of SISPA is available at https://www.bioconductor.org/packages/SISPA/.
Archived source code as at the time of publication: https://doi.org/10.5281/zenodo.1164284 (Dwivedi & Kowalski, 2018)
License: shinySISPA is available under the GNU public license (GPL-3)
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)