Introduction
Psychiatric disorders, including autism, schizophrenia, bipolar disorder, attention deficit hyperactivity disorder (ADHD) and depression, affect millions of people and are a major socio-economic burden1–3. The identification of underlying genetic defects and risk factors is becoming increasingly efficient because of genome-wide interrogation methodologies, yet owing to the complex multifactorial origin of most cases, a conclusive molecular diagnosis is made for only a minority of patients. Therefore, the underlying causes for these conditions are poorly understood, and often treatment is still based on symptomology4–6. In 2003, Rubenstein and Merzenich proposed autism spectrum disorders (ASDs) to be caused by an increase in the ratio between excitation and inhibition, called the E/I balance7. Since then, this hypothesis has been substantiated by a vast number of studies and also has been implicated in other psychiatric disorders such as schizophrenia8, consistent with their partially overlapping phenotypes9. Recently, the focus has shifted to changes to the inhibitory side of the E/I balance10,11, in particular to one class of inhibitory neurons, parvalbumin (PV)-positive interneurons12. In this review, we focus on the role of the inhibitory system in psychiatric disorders and explore the changes to the inhibitory systems in different disorders. We then discuss the role and function of PV interneurons and highlight the changes to this specific class of interneurons in the various psychiatric disorders.
Evidence for inhibitory dysfunction in psychiatric disorders
Since Rubenstein and Merzenich postulated their hypothesis of a reduced E/I balance in ASDs, there has been an increasing amount of evidence for disrupted inhibitory control in psychiatric disorders. This evidence comes from post-mortem studies and studies of patient phenotypes.
Firstly, post-mortem studies on patient brains have revealed consistent changes to the inhibitory system in various disorders. Studies of autistic brains revealed reduced expression of the gamma-aminobutyric acid (GABA) synthesizing enzymes GAD65 and GAD67, as well as various GABA receptor subunits, in parietal cortex and cerebellum13,14. In schizophrenia, reductions of interneuron markers have been found in the prefrontal cortex15–18, a region strongly implicated in this condition19. Interestingly, in recent years, this reduction has been shown to be caused by a reduction of the expression of the interneuron markers rather than a reduction of the number of interneurons20–22, which indicates reduced activity of these neurons23–25. In addition, both increased and decreased numbers of specific interneuronal subtypes are reported in bipolar disorder17,26, while a reduced inhibitory function is reported in depression27,28 and bipolar disorder29.
Secondly, patients with psychiatric disorders display phenotypes that are strongly correlated to impaired inhibition. Epilepsy is a common comorbidity with psychiatric disorders and has consistently been linked to impaired inhibitory function30–33. In patients with autism, it is estimated that the prevalence of epilepsy comorbidity is around 25%34,35. However, this is dependent on the type of autism, and the prevalence can be as high as 80% in Rett syndrome36, a monogenic form of autism caused by mutation in the MeCP2 gene37. It is currently unclear whether schizophrenia is a risk factor for epilepsy. A limited number of studies have been dedicated to this question, and contradicting results have been reported38,39. However, patients with epilepsy show an increased risk of schizophrenia or schizophrenia-like psychosis40. Likewise, patients with epilepsy show an increased risk for ADHD41,42.
Another recurrent phenotypic change is the altered power of gamma oscillations, as measured with electroencephalography or magnetoencephalography in humans, indicating changes in neuronal synchrony43. Gamma oscillations are important for integration of information in neuronal circuits and have been linked to various functions, including attention44 and memory45. It was shown that PV-positive interneurons46, specifically PV-positive basket cells (see below), play an important role in these osciliations43,47,48. Changes in gamma oscillations are consistently found in patients with schizophrenia49, affecting different regions, including the prefrontal cortex50,51. Interestingly, while a decrease in gamma power is linked to negative symptoms of this disorder, such as psychomotor poverty52, increased gamma power has been observed during positive symptoms, such as hallucinations53. In addition, computational studies suggest a central role for inhibitory synaptic scaling in maintaining a stable neuronal network54 and found changes in inhibitory transmission to be sufficient to explain the changes in gamma oscillations in schizophrenia55. Together, altered inhibitory control is believed to lead to a change in the power of gamma oscillations, which play a central role in schizophrenia56.
Though studied mainly in schizophrenia, changes in gamma oscillations have been observed in other psychiatric disorders, including autism, ADHD and bipolar disorder57–63. For example, children with autism show a reduced gamma frequency modulation to a visual task64, whereas in ADHD, increased power and synchrony were observed59–62. Together, post-mortem and patient studies point to an important role for altered inhibitory function in various psychiatric disorders and indicate a vital role for inhibition in the maintenance of the E/I balance in the healthy brain.
The central role of parvalbumin-positive interneurons in E/I balance
Cortical and hippocampal synaptic inhibition is mediated by inhibitory interneurons, most of which use GABA as their neurotransmitter. While interneurons make up around 20% of the total neuronal population, they are highly diverse65,66. For example, different classes of interneurons are specialized to target the dendrites, soma or axon initial segment (AIS) of pyramidal neurons65. This large variety of cell types is believed to illustrate the distinct functions that these cells have in regulation of the network67. Cortical interneurons can be segregated in three non-overlapping groups by means of specific markers: PV, somatostatin (SOM) and the serotonin receptor 3a (5HT3aR), accounting for 40%, 30% and 30% of the total interneuron population, respectively68. 5HT3aR-positive cells mainly originate from the caudal ganglionic eminence and are further divided as vasoactive intestinal peptide (VIP)-positive and VIP-negative interneurons68. VIP-positive interneurons mainly inhibit other interneurons and play an important role in disinhibition of the local circuit69, where they receive excitatory input from other cortical areas70,71. VIP cells mainly inhibit SOM cells72 but also target PV interneurons23 and are involved in the regulation of the behavioural state of the network71,73. Recent studies have suggested a direct inhibition by VIP interneurons of pyramidal cells in cortex74,75. Despite the prominent, mainly disinhibitory, function of VIP cells in the network, only a limited number of studies have implicated VIP cells to be involved in psychiatric disorders26,76.
SOM interneurons are a diverse class of interneurons originating from the medial ganglionic eminence (MGE)68. These interneurons target non-SOM interneurons72 as well as the dendritic domain of pyramidal neurons, including dendritic spines77. SOM interneurons regulate the integration of local excitatory input78,79 and have been shown to regulate synaptic plasticity via the control of dendritic calcium spikes in pyramidal cells, affecting learning tasks80. Increasing evidence implicates SOM interneurons in psychiatric disorders. Disinhibition of SOM interneurons leads to an anti-depressive–like phenotype in mice81, and reduced levels of SOM in cerebral spinal fluid have been linked to major depression and mood disorders82. In addition, a recent article shows a role for SOM interneurons in gamma oscillations in the visual cortex83, hinting towards a possible role for SOM interneurons in the changes in gamma oscillations observed in psychiatric disorders.
PV interneurons are MGE-derived and are electrophysiologically identified by their fast-spiking phenotype. Although PV interneurons make up only a small part of the entire neuronal population84,85, these interneurons are strongly implicated in psychiatric disorders and have been shown to play an important role in the regulations of the E/I balance8,10,86. PV interneurons are involved in gamma oscillations (see above), and various mutations in disease-linked genes affect PV interneuron function (discussed below) (Table 1). Different subtypes of PV interneurons are distinguished: basket cells, chandelier cells, bistratisfied cells, and, in hippocampus, oriens-alveus-lacunosum-moleculare cells87,88 form the largest of these groups, the first two of which are most widely studied. Here, we will discuss both types and focus on their respective roles in the network.
Table 1. Genes linked to psychiatric disorders affect distinct subcellular aspects.
| Aspect | Gene | Syndrome/Disorder | Model | Investigated
region | Phenotype | Reference |
|---|
| Input | Erbb4 | SZ | PV interneuron KO | Hippocampus | Reduced excitatory input to
PV basket cells and chandelier cells | 120 |
| Nrg1 | SZ | NRG1 treatment of
dissociated cortical
cultures | Cortical
(cultures) | Increased excitatory synapse
number onto interneurons | 121 |
| Fmr1 | Fragile X syndrome;
ASD | Fmr1 KO mouse | Cortex | Reduced local excitatory input
onto FS interneurons | 122 |
| DISC1 | SZ, ASD, depressive
disorder, BD | PV-specific shRNA
KD in vivo | Cortex | Increased excitatory input onto PV
interneurons | 123 |
| Nlgn3 | ASD | PV interneuron KO | Hippocampus
Hippocampus | Decreased NMDAR responses
Increased glutamate release onto
PV interneurons | 124 |
| Mecp2 | Rett syndrome; ASD | PV interneuron KO | Cortex | Reduced local excitatory input
onto PV interneurons | 125 |
| Intrinsic | Mecp2 | Rett syndrome; ASD | PV interneuron KO | Cortex | Increased intrinsic excitability of
PV interneurons | 125 |
| Dysbindin | SZ | Dysbindin KO mouse | Cortex | Reduced excitability of FS
interneurons | 126 |
| Scn1a | Dravet syndrome;
ASD | Scn1a KO mouse | Hippocampus | Impaired action potential kinetics
in interneurons | 127 |
| Shank3 | ASD, SZ | Shank3B KO mouse | Cortex, Striatum | Reduced activity of PV
interneurons | 22 |
| Output | Erbb4 | SZ | PV interneuron KO | Hippocampus | Reduced cartridges from
chandelier cells onto pyramidal
neurons | 120 |
| Nrg1 | SZ | Overexpression in
pyramidal neurons | Cortex | Increased basket cell and
chandelier cell boutons onto
pyramidal neurons | 128 |
| Tsc1 | Tuberous sclerosis;
ASD | Sparse Tsc1 deletion
in CA1 pyramidal
neurons | Hippocampus | Reduced inhibitory synaptic
strength onto pyramidal neurons | 129 |
| Ube3a | Angelman syndrome;
ASD | Maternal loss of
Ube3a mouse | Cortex | Reduced inhibitory drive from FS
and non-FS interneurons onto
pyramidal neurons | 130 |
| Shank3 | ASD, SZ | Shank3-exon9 KO
mice | Cortex | Reduced inhibitory input onto
pyramidal neurons | 131 |
| | Shank3-exon9 KO
mice | Hippocampus | Increased inhibitory input onto pyramidal neurons | 131 |
| Git1 | ADHD | Git1 KO mouse | Hippocampus | Reduced inhibitory inputs onto
pyramidal neurons | 132 |
| Cdh13 | ADHD | Cdh13 KO mouse | Hippocampus | Increased number of inhibitory
synapses onto pyramidal neurons | 133 |
| Nlgn 2 | ASD | Nlgn2 KO mouse | Cortex | Reduced inhibitory drive onto
pyramidal neurons from FS
interneurons | 134 |
| ASD | Nlgn2 KO mouse | Hippocampus | Reduced number of perisomatic
synapses onto pyramidal neurons | 135 |
| Nlgn 3 | ASD | Nlgn3 R451C mouse | Hippocampus | Reduced inhibitory drive from
PV basket cells onto pyramidal
neurons | 136 |
| Cntnap2 | ASD | shRNA KD in
dissociated cortical
cultures | Cortical
(cultures) | Reduced inhibitory drive onto
pyramidal neurons | 137 |
PV basket cells
Basket cells are the largest group of PV interneurons and specifically target the soma and proximal dendrite of pyramidal neurons87. The perisomatic location of these axon terminals allows PV basket cells to have a strong control over the excitability of pyramidal neurons. Among other cortical inputs, PV basket cells receive the same excitatory input as their pyramidal cell targets, wiring the basket cell into a feed-forward circuit: excitatory input will excite both the PV basket cell and the pyramidal neuron, followed by the PV basket cell inhibiting the pyramidal neuron. The delay between the excitatory and inhibitory input onto the pyramidal cell creates a coincidence detection window, in which excitatory input can summate to elicit an action potential in the pyramidal cell89. If inhibitory input arrives at the pyramidal cell before an action potential is evoked, the somatic targeted GABA action will prevent action potential initiation. So PV basket cells allow action potential initiation in pyramidal neurons only if the excitatory information is time-locked and of sufficient strength.
In order to mediate fast inhibition, PV basket cells are optimized for fast signalling85. Action potentials are initiated at the AIS and propagate at high velocities through the axon90 which is enriched for the fast sodium channel NaV1.191. Synaptically, calcium inflow is mediated by fast P/Q-type calcium channels92, which are located directly adjacent to the release site93. The post-synaptic site, on the pyramidal neuron, contains the fast GABAAα1 receptor subunit94. These fast properties ensure an optimal speed of PV basket cell signalling and tightly regulate coincidence detection windows of pyramidal neurons.
Chandelier cells
Chandelier cells, or axo-axonic cells, are a group of interneurons that target the AIS of pyramidal neurons95,96. These cells form vertically oriented clusters of axon terminals, called cartridges, giving them a chandelier-like appearance. A single pyramidal cell receives contacts from multiple chandelier cells97, forming an average of 3 to 5 boutons each98 depending on the brain region99 and age100. The synapses are enriched for the GABAAα2 receptor subunit101,102 and pre-synaptically express the high-affinity GABA transporter 1 (GAT1)103. The function of chandelier cells in the network remains largely unknown. However, chandelier cell activity has been shown to increase with increasing network activity104. In addition, the axon terminals of chandelier cells are specifically absent from the epileptic focus105,106, and pharmacological induction of seizures leads to a loss of chandelier cells in rats107, suggesting a role in preventing excessive excitatory activity in the network for these interneurons104.
Since their discovery, there has been a debate about the actions of chandelier cells (reviewed by Wang and colleagues98). Some studies using brain slice recordings showed a depolarizing108 and even excitatory109 action for these interneurons, whereas others report an inhibitory action110. However, chandelier cell membrane potential fluctuations resembling in vivo patterns appear strongly inhibitory111, and recordings in vivo also suggest an inhibitory role for these interneurons112,113.
Whereas patient studies of schizophrenia patients consistently identify both a reduction in the number or length of chandelier cell cartridges114,115 as well as the misregulation of proteins associated with chandelier cell synapses12,116–118, mouse research on this interneuron class is hampered by the absence of a strategy to specifically target these interneurons. As a result, a limited number of studies focus on chandelier cells but instead report on PV interneurons in general, or PV basket cells, which provide a more accessible target of study because of their relative abundance (Figure 1). Nonetheless, chandelier cells are considered to play an important role in psychiatric disorders11,98,119, and the development of strategies to specifically target this interneuron subtype would be an important step towards understanding the role of these interneurons.
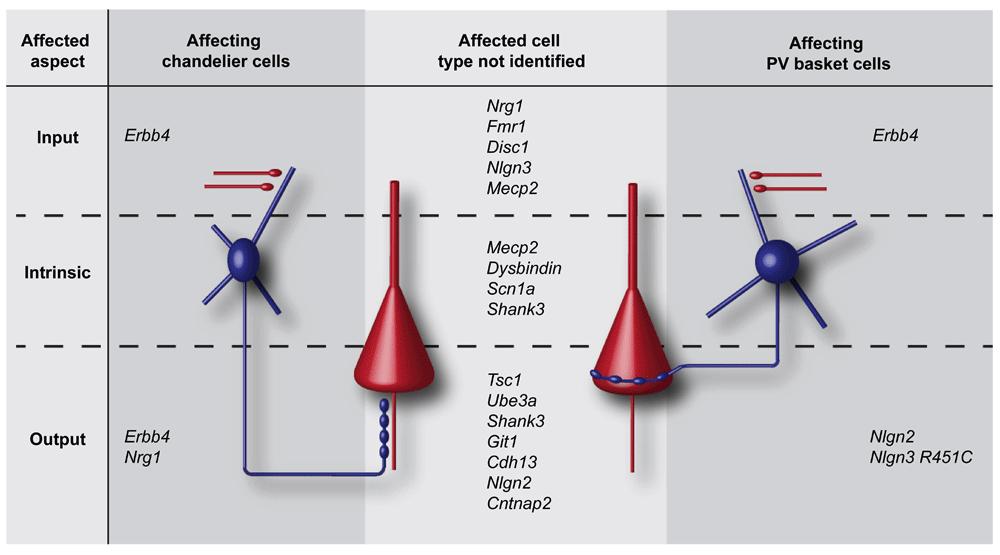
Figure 1. Genes linked to psychiatric disorders affect inhibition on different subcellular aspects.
(Left) Genes affecting the input, intrinsic properties or output of chandelier cells. (Right) Genes affecting the input, intrinsic properties or output of parvalbumin-positive (PV) basket cells. (Middle) Genes affecting input, intrinsic properties or output of interneurons, without the interneuron subtype being identified. Pyramidal cells are shown in red, and interneurons are shown in blue.
Targeting the perisomatic region (basket cells) or AIS (chandelier cells) gives these interneurons strong control over pyramidal cell excitability, and regulation of the synaptic strength of these interneurons is important for normal function of the network. For example, Xue and colleagues have shown that pyramidal neurons receive an amount of synaptic inhibition that is proportional to the amount of synaptic excitation they receive138, maintaining the E/I balance on the pyramidal cell. Manipulation of pyramidal cell activity leads to a compensatory change in inhibitory drive onto these cells, specifically from PV interneurons138. In addition, PV interneuron activity is reduced during learning and increased during fear conditioning, and an experimental increase of PV interneuron activity leads to impaired learning23. Apart from synaptic connections, basket cells139 and chandelier cells111,140 connect via electrical connections, called gap junctions. This electrical coupling synchronizes the interneurons139, which in turn allows them to synchronize the network141 (for example, in gamma oscillations142).
Together, these studies indicate that control of the E/I balance by PV interneurons is important for normal network function and that PV interneuron-mediated inhibition can be regulated upon alterations in the network state. Changes in PV interneuron-mediated inhibition would shift the E/I balance and lead to a disruption in network function. Indeed, various parameters affecting inhibitory function of PV cells are consistently found to be altered in psychiatric disorders. In the next section, we focus on studies on animal models of these conditions and discuss how different changes affecting PV cell activity lead to a shift of the E/I balance (Table 1).
Altered PV interneuron activity is caused by changes to different subcellular aspects
Alterations to the inhibitory drive, affecting the E/I balance, can arise in different ways. Reduced excitatory input onto interneurons, reduced intrinsic excitability of interneurons, and a reduction in inhibitory synapse number or strength onto pyramidal cells all result in a shift of the E/I balance towards excitation. Indeed, patient and animal studies of psychiatric disorders consistently report changes affecting inhibitory function. A complicating factor in the interpretation of these results comes from the dynamic ability of neuronal networks to adapt to changes, known as homeostatic plasticity. Homeostatic plasticity is the ability of neurons to maintain their levels of excitability within a narrow range and is a constantly active feedback process143. For example, classic experiments have shown that blocking of action potentials leads to a strengthening of excitatory144 and a weakening of inhibitory synapses145. Apart from their synaptic input, neurons can regulate their intrinsic excitability146, which is observed both in excitatory and inhibitory neurons in culture147 and in vivo148. This means that genetic mutations affecting a specific neuronal property in a specific cell type can trigger homeostatic processes affecting other properties or cell types. In this way, mutations affecting both inhibitory or excitatory cells could ultimately affect inhibitory function86. It is therefore difficult to distinguish the direct effect of a gene related to a psychiatric disorder from the network adaptation it causes.
Nonetheless, changes to the function of PV interneurons, either direct or indirect, are consistently observed in psychiatric disorders affecting input, output and intrinsic properties, which all lead to an altered inhibitory action of these cells onto their targets (Figure 1).
Changes to the input onto PV interneurons
Excitatory inputs onto neurons drive their excitability. Depending on the brain region, PV interneurons receive various types of excitatory input85, and the amount of excitatory inputs onto PV interneurons is dynamically regulated by behaviour23. Changes in the amount of excitation onto PV interneurons, altering their activity, have been reported in mouse models of various psychiatric disorders.
The tyrosine kinase receptor ErbB4 has been identified as a risk gene for schizophrenia in genome-wide association studies (GWASs)149,150. In the adult mouse, expression of ErbB4 is restricted to interneurons128,151 and localizes to both the axon terminals128,152 and post-synaptic densities128,151. Selective removal of ErbB4 from PV interneurons causes a reduction in excitatory synapses formed onto both PV basket cells and chandelier cells as well as a reduced number of PV synapses formed on pyramidal neurons120,121. This reduced input and output connectivity of PV interneurons is indicative of a reduced inhibitory drive onto pyramidal neurons. As a result of this reduced inhibition of the pyramidal neurons, these neurons become more active, as was seen from the increased frequency of excitatory inputs to both pyramidal neurons and PV interneurons120. Consequently, recording of the local field potential in vivo revealed a hyperactive network and increased gamma oscillations120. Single-nucleotide polymorphisms in neuregulin 1 (NRG1), a ligand of ErbB4, have been implicated in schizophrenia153 and bipolar disorder154,155. Treatment of neuronal cultures with NRG1, activating ErbB4, leads to an increase in excitatory synapses formed onto interneurons121. Together, these data show that ErbB4 signalling plays an important role in the regulation of excitatory synapse number onto PV interneurons and that disruption of this system leads to a shift of the E/I balance towards excitation120.
Also, studies on animal models of ASD have reported postsynaptic changes on PV interneurons. Fragile X syndrome is caused by reduced or absent levels of the RNA-binding protein FMRP, leading to intellectual disability and, in about half of the affected males, ASD156,157. While changes to long-term depression on excitatory cells have been the centre of attention for this condition158, changes to the inhibitory system have consistently been identified159,160. Fmr1 knockout (KO) mice show a reduced expression of GABAA receptor subunits161 as well as a reduction in the number of PV interneurons162. In addition, these mice show a marked reduction in local, but not thalamic, excitatory input onto fast-spiking interneurons in layer 4 of the somatosensory cortex, while both the connectivity of fast-spiking interneurons onto pyramidal neurons and excitatory inputs onto pyramidal neurons were unaltered122. Consistently, the resulting reduced inhibitory drive from fast-spiking interneurons was accompanied by reduced synchrony of gamma oscillations122. These changes point towards an altered E/I balance towards excitation in fragile X syndrome163.
Mutations in another gene linked to autism, the transcriptional modulator methyl-CpG-binding protein 2 (MECP2), the causative gene for Rett syndrome37, show a similar phenotype. Selective removal of Mecp2 from PV cells leads to a specific reduction of local excitatory input, but not thalamocortical input, onto these cells in layer 4 of the visual cortex at post-natal day (P) 30125. In addition, experimentally evoked PV interneuron input onto pyramidal cells was unaltered125. Calcium imaging revealed that these synaptic changes lead to a reduced visually evoked, but not spontaneous, activity of PV interneurons125. Paired recordings at P15 revealed an increased inhibition from PV interneurons through an earlier maturation in Mecp2 KO mice164, and this earlier maturation might influence network development through interference with the normal critical period164,165, leading to the changes observed at later ages. This notion is consistent with the recent idea that developmental changes might play an important role in the development of psychiatric symptoms later in life166. The exact contribution of PV interneurons to the phenotypes observed in Rett syndrome is still unclear. While a general interneuron removal of Mecp2 does recapitulate most Rett syndrome phenotypes167, studies removing Mecp2 specifically from PV interneurons have been able to replicate only some of these phenotypes168 or none at all125. Of note, selective removal of Mecp2 from SOM interneurons also recapitulates part of the Rett syndrome phenotypes168, and selective removal of Mecp2 from either PV interneurons or SOM interneurons has been reported to cause circuit-wide deficits in information processing169. From these studies, a picture is emerging in which excitatory inputs onto PV interneurons are found to be altered in different psychiatric disorders, leading to a reduced activity of these neurons and thereby tilting the E/I balance towards excitation.
Changes to the intrinsic properties of PV interneurons
The intrinsic properties of neurons are important in translating input into output. Altering these properties allows the neurons to regulate their excitability170,171, through which they play an important role in the maintenance of the E/I balance143. Whereas some of these changes might be causative to psychiatric disorders, others are believed to be compensatory. For example, selective deletion of Mecp2 from PV interneurons leads to an increased membrane potential, as well as a hyperpolarized action potential threshold in these cells at P30125, but only a slight hyperpolarization of the membrane potential at P15164. These changes increase the cell’s excitability and are most likely compensatory for the reduced excitatory synaptic input described above125 but could still act towards deficits in information processing observed in these animals169.
Family-based association data have identified dysbindin (DTNBP1) as a susceptibility gene for schizophrenia172, whose dominant circuit impact is impaired inhibition173. Dysbindin KO mice show a reduced number of PV interneurons in hippocampal CA1173,174, and transcriptome changes of various proteins involved the regulation of intrinsic properties174. Recordings from PV interneurons in dysbindin KO mice show a reduction in action potential frequency resulting in a reduced inhibitory drive126. Interestingly, dopamine D2-receptor expression is increased in these mice, and application of the D2-receptor antagonist quinpirole increases PV interneuron action potential frequency more in dysbindin KO mice than in wild-type mice, suggesting that the changes in action potential frequency are compensatory126.
Intrinsic changes can also be the primary cause of psychiatric disorders. Single-gene mutations in SCN1A, encoding the sodium channel NaV1.1α subunit, give rise to Dravet syndrome, a rare genetic epileptic encephalopathy. Patients with Dravet syndrome suffer from epilepsy and have an increased risk for autism175,176. NaV1.1 is enriched in the AIS of inhibitory neurons177, primarily of PV interneurons91, where axon potentials are initiated178. Interneurons from Scn1a heterozygous and KO mice show reduced firing frequencies to current injections as well as a reduced action potential amplitude and an increased action potential width, indicating a reduced inhibitory control over downstream targets127. Removal of Scn1a specifically from forebrain interneurons179 or PV interneurons specifically180 recapitulates phenotypes found in patients. In addition, increasing GABA signalling by application of the positive allosteric GABAA receptor modulator clonazepam was sufficient to rescue the abnormal social behaviour in Scn1a+/− mice181. These data show that loss of SCN1A primarily affects interneurons and that the consequently reduced inhibitory drive plays an important role in Dravet syndrome.
Recently, a new hypothesis has been proposed in the field of schizophrenia, focussing on the myelination of PV interneurons as a point of pathological convergence182. As discussed above, PV cells play a central role in schizophrenia. Myelination abnormalities, including white matter abnormalities183, reduced numbers of oligodendrocytes184 and post-mortem gene expression analysis185, have been identified in schizophrenia. Myelination of PV interneurons has been observed in rats186, mice187 and post-mortem in humans188. Myelination is important for fast action potential propagation189, and deficits in the myelination of PV interneurons are proposed to disrupt inhibitory network function182. However, this appealing hypothesis remains to be experimentally tested.
Changes to synapses formed by PV interneurons
Changes to PV synapses are abundantly studied and identified in various conditions. Post-mortem studies of schizophrenia patients consistently identify changes to the cartridges formed by chandelier cells, showing a decrease in pre-synaptic GAT1 expression116,117 and an increase in the expression of post-synaptic GABAAα2118. These changes would lead to an increased inhibitory drive from chandelier cells and are believed to be compensatory for a reduced activity of these cells, as indicated by reduced levels of GAD67 in PV cells190. In addition, a recent study shows a reduction in the density of a specific type a cartridges, calbindin-positive cartridges, in schizophrenia114.
Besides changes in the input to PV interneurons, mutations in Erbb4 also lead to a reduction in synapses formed by PV interneurons on pyramidal neurons, specifically from chandelier cells120,128. In addition, overexpression of Nrg1, the ligand for ErbB4, in pyramidal neurons increases bouton density on both the AIS and the soma of pyramidal neurons. The increase in bouton density on the AIS originates from chandelier cells since only these cells target the AIS. The origin of the increase in perisomatic bouton density is not clear since a recent report has shown that synapses formed by cholecystokinin (CCK) basket cells require ErbB4 function to form perisomatic synapses191. Future research should unveil whether the increase in perisomatic boutons density arises from PV or CCK basket cells.
Tuberous sclerosis is a disorder whose symptoms include epilepsy and autism192. Loss-of-function mutations in the mammalian target of rapamycin (mTOR)-negative regulators TSC1 or TSC2 underlie this condition193. Slice recordings from TSC1 KO neurons revealed a reduced inhibitory drive onto CA1 pyramidal neurons, while excitatory input was unaltered129. TSC1 KO neurons were created by sparsely targeting neurons in a conditional TSC1 KO mouse with a cre-expressing adeno-associated virus129. The finding that sparse KO of TSC1 leads to a similar phenotype as in neurons from full KO animals indicates that these results are cell-autonomous rather than compensatory129.
Angelman syndrome, caused by loss-of-function mutations or deletion of E3 ubiquitin ligase UBE3A194, is characterized by epilepsy and autism195,196. Mouse models for Angelman syndrome recapitulate human phenotypes197 and initially were found to show a reduced excitatory drive onto pyramidal neurons198. However, inhibitory input was also found to be decreased, caused by defects in synaptic vesicle cycling130. It was hypothesized that the reduced inhibition could outweigh the reduced excitation, leading to the epilepsy and autism phenotypes obeserved130,199. Consistent with this idea, a recent study using in vivo whole cell recordings shows that pyramidal neurons in Ube3a KO mice display decreased orientation selectivity200, indicative of reduced inhibition201,202. However, these mice also show increased excitability of pyramidal neurons, which is non–cell-autonomous, suggesting that pyramidal neurons homeostatically increase their excitability because of a relative decrease of excitation200. While it is unclear whether reduced inhibition or reduced excitation has a stronger impact on pyramidal neurons in Angelman syndrome, the change in their relative contribution, resulting in an altered E/I balance, seems to play a pivotal role in this condition.
Single SHANK3 mutations, at the level of point mutations or microdeletions, have been identified in patients with ASD203. In the human genome, there are three SHANK genes (SHANK1-3), which all code for scaffold proteins located at the postsynaptic density of excitatory synapses, of which SHANK3 is best studied204–206. Because of this localisation, most studies have focussed on excitatory synapses, where Shank3-deficient mice show reduced cortico-striatal connectivity207, impaired long-term potentiation208 and reduced GluA1 expression209. Recent studies, however, indicate that the inhibitory system is also affected. Shank3 mutant mice lacking exon 9 show reduced inhibitory input onto layer 2/3 pyramidal neurons but an increase of these events in CA1 pyramidal neurons131. In addition, PV levels are reduced in Shank1 KO and Shank3b KO mice, indicating reduced activity22.
Presynaptic neurexins and their postsynaptic partners neuroligins are a large class of cell-adhesion molecules that have been shown to play important roles in synaptic specificity210. Overexpression or knockdown of neuroligins leads to an increase or decrease in synapse number, respectively211. Neuroligins are expressed at specific synapses: neuroligin 1 (NLGN1) is mainly expressed at excitatory synapses212, NLGN2 is expressed at inhibitory synapses213, NLGN3 is expressed at both inhibitory and excitatory synapses214 and NLGN4 is expressed at glycinergic synapses215. Mutations and deletions affecting human NLGN3, including a gain-of-function mutation, and NLGN4 de novo mutations have been found in Swedish families and have been associated with autism216. Nlgn3 deletion in mice leads to increased inhibitory transmission onto pyramidal neurons217, specifically from CCK-positive interneurons because of an increased tonic endocannabinoid signalling, whereas PV interneuron connectivity is unaffected136. A recent study, however, has shown that conditional deletion of Ngln3 from PV interneurons alters AMPA/NMDA (alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid/N-methyl-d-aspartate) ratio of excitatory input onto these cells and causes reduced gamma oscillations124. In addition to the loss of NLGN3, a gain-of-function amino acid substitution (R451C) in NLGN3 is associated with autism216. Mice carrying this mutation show a strong reduction in inhibitory drive from PV interneurons while increasing the inhibitory drive from CCK cells136.
Nlgn4 KO mice show a reduced number of perisomatic inhibitory synapses in hippocampus and a concomitant reduction in inhibitory input218. In addition, a reduced power of evoked gamma oscillations in acute slices was observed218. Different mutations in NLGN2 have been linked to autism219 and schizophrenia220. Deletion of Nlgn2 has been shown to specifically reduce the amount of perisomatic synapses on pyramidal neurons in hippocampus135 and reduce inhibitory transmission from PV interneurons but not SOM interneurons134.
Neurexins are less well studied in the context of psychiatric disorders. There are three neurexin genes, each coding for an α- and β-neurexin. Mutations in neurexin (NRXN1) have been associated with autism221 and schizophrenia, affecting both NRXN1α and NRXN1β222,223. Nrxn1α KO mice do not show changes in inhibitory drive but do show a reduced excitatory drive onto CA1 pyramidal neurons224. However, mice carrying mutations in Nrxn1β show a reduced frequency of both inhibitory and excitatory input onto cortical pyramidal neurons225 suggestive of a reduced number of synaptic contacts. These studies show that neuroligin and neurexin mutations that are linked to autism affect the inhibitory system, including perisomatic-targeting interneurons.
Studies of ADHD rodent models have mainly focussed on the dopamine system and excitatory synapses226. However, recent studies identify changes to inhibitory connectivity. G protein–coupled receptor kinase interacting protein 1 (GIT1) has been identified by a GWAS as a risk gene for ADHD132, but recent studies challenge this claim227,228. Git1 KO mice show ADHD-like phenotypes, including hyperactivity132. While excitatory input to CA1 pyramidal neurons remains unaltered, the frequency of inhibitory inputs is reduced, suggesting a reduction of inhibitory synaptic contacts132, consistent with previous studies showing a role for GIT1 in inhibitory synapses229. In addition, PV expression was reduced while other interneuron markers remained unaltered132.
Another ADHD-linked gene identified by GWASs, cadherin 13 (CDH13)230–232, is exclusively expressed in inhibitory neurons133. Cdh13 KO mice show an increase in inhibitory synaptic contacts onto CA1 pyramidal neurons, while excitatory inputs remained unaltered133. This increase in inhibitory synaptic contacts could underlie the increase in gamma oscillations observed in ADHD patients discussed before59–62. Cdh13 KO mice show a reduced number of interneurons at embryonic day 18.5233 but not at P21133. Experiments are needed to test whether the changes in interneuron number have a role in the aetiology of ADHD.
In addition to changes in the number and strength of inhibitory synapses, changes are found in the modulation of inhibitory synaptic transmission. Metabotropic GABA receptors, GABAB receptors, are expressed both pre- and postsynaptically in GABAergic synapses234. Postsynaptic GABAB receptors activate potassium channels that hyperpolarize the postsynaptic cell235. Presynaptic GABAB receptors, in addition, can reduce calcium inflow and reduce neurotransmitter release236,237. A reduction of GABAB subunit expression has been observed in post-mortem studies of patients with schizophrenia238,239, patients with bipolar disorder240 and patients with major depression241 as well as animal models for schizophrenia242–244. However, more research is required to identify the exact role for GABAB receptors in psychiatric disorders.
In summary, changes to the inhibitory drive of PV interneurons can be caused by changes affecting the input, output or intrinsic properties of PV interneurons, and animal models of various psychiatric disorders all show alterations to one or more of these aspects, tilting the E/I balance.
Conclusions
Psychiatric disorders are a diverse group of disorders, but changes to the inhibitory system seem to be a point of convergence. Impairment of normal inhibitory function can arise from input to, output from, or intrinsic properties of inhibitory neurons. Altered inhibitory activity or drive leads to changes in signal processing, which in turn is believed to underlie the phenotypic changes observed in the various psychiatric disorders. PV-positive interneurons play a pivotal role in these conditions, possibly through their strong inhibitory effect on pyramidal cell activity due to the axonal or perisomatic targeting of their axons in combination with the nature of their functions in the network.
Changes to either excitation or inhibition will change the ratio between these two types of input, leading to a change in the E/I balance. The examples described above indicate that various psychiatric disorders occur following changes to the input, output or intrinsic properties of specific interneurons, PV interneurons, leading to an altered activity of these neurons. It seems from the studies discussed that specific changes in the E/I balance lead to a disruption of specific function(s) in the network that affect signal processing in a specific way to result in a specific psychiatric phenotype. This might explain why different disorders present different phenotypes, in both patients and animal studies. For example, changes in chandelier cell cartridges seem to be more prominent in schizophrenia. However, other factors are likely to contribute to the development of a specific disorder. Apart from the affected PV cell type, the direction of the altered activity (increased/decreased E/I balance) might be an important factor. Another interesting aspect might be the changes in interneuron-interneuron connectivity, which could lead to altered signal integration and network activity. Some genes have been associated with multiple psychiatric disorders, indicating that a mutation does not in all cases lead to a specific condition. Individual difference in compensatory plasticity could subtly affect network development, steering the developing network towards a specific disorder. It should be noted that while homeostatic changes might compensate a specific alteration, this compensation might disrupt other pathways. While the above-mentioned factors potentially all play a role in the development of specific psychiatric disorders, more research is needed to identify how specific alterations to PV interneurons affect network processing and behaviour.
The notion that psychiatric disorders are caused by changes to the inhibitory drive from PV interneurons means that a restoration of this drive could improve patient symptoms. An interesting possibility of counteracting the changes to the inhibitory system would be to make use of the neuron’s ability for homeostatic plasticity170. Homeostatic mechanisms function to keep neurons in an optimal range of activity245. Understanding which genetic and molecular mechanisms underlie these homeostatic processes opens the possibility to ‘hijack’ these pathways and manipulate the neuron’s activity in a way to compensate for the altered inhibitory drive. In this way, it might be possible to selectively up- or downregulate the activity levels of specific interneuron populations and thereby ameliorate the symptoms observed in patients with the various disorders.
While psychiatric disorders are still far from being fully understood, the study of the role of the inhibitory system in these conditions might further increase our understanding of both the diseased and the healthy brain and hopefully could lead to treatment or alleviation of the symptoms for those suffering from these conditions.
Competing interests
The authors declare that they have no competing interests.
Grant information
The author(s) declared that no grants were involved in supporting this work.
Acknowledgements
The authors would like to thank Wei Ba and Clémence Bernard for valuable discussions.
Faculty Opinions recommendedReferences
- 1.
Gore FM, Bloem PJ, Patton GC, et al.:
Global burden of disease in young people aged 10–24 years: a systematic analysis.
Lancet.
2011; 377(9783): 2093–102. PubMed Abstract
| Publisher Full Text
- 2.
Lee FS, Heimer H, Giedd JN, et al.:
Mental health. Adolescent mental health--opportunity and obligation.
Science.
2014; 346(6209): 547–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 3.
Elsabbagh M, Divan G, Koh YJ, et al.:
Global prevalence of autism and other pervasive developmental disorders.
Autism Res.
2012; 5(3): 160–79. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 4.
Remington G, Foussias G, Fervaha G, et al.:
Treating Negative Symptoms in Schizophrenia: an Update.
Curr Treat Options Psychiatry.
2016; 3(2): 133–50. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 5.
Doyle CA, McDougle CJ:
Pharmacologic treatments for the behavioral symptoms associated with autism spectrum disorders across the lifespan.
Dialogues Clin Neurosci.
2012; 14(3): 263–79. PubMed Abstract
| Free Full Text
- 6.
van Os J, Kapur S:
Schizophrenia.
Lancet.
2009; 374(9690): 635–45. PubMed Abstract
| Publisher Full Text
- 7.
Rubenstein JL, Merzenich MM:
Model of autism: increased ratio of excitation/inhibition in key neural systems.
Genes Brain Behav.
2003; 2(5): 255–67. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 8.
Gao R, Penzes P:
Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders.
Curr Mol Med.
2015; 15(2): 146–67. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 9.
Canitano R, Pallagrosi M:
Autism Spectrum Disorders and Schizophrenia Spectrum Disorders: Excitation/Inhibition Imbalance and Developmental Trajectories.
Front Psychiatry.
2017; 8: 69. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 10.
Marín O:
Interneuron dysfunction in psychiatric disorders.
Nat Rev Neurosci.
2012; 13(2): 107–20. PubMed Abstract
| Publisher Full Text
- 11.
Lewis DA, Hashimoto T, Volk DW:
Cortical inhibitory neurons and schizophrenia.
Nat Rev Neurosci.
2005; 6(4): 312–24. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 12.
Lewis DA, Curley AA, Glausier JR, et al.:
Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia.
Trends Neurosci.
2012; 35(1): 57–67. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 13.
Fatemi SH, Reutiman TJ, Folsom TD, et al.:
GABAA receptor downregulation in brains of subjects with autism.
J Autism Dev Disord.
2009; 39(2): 223–30. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 14.
Fatemi SH, Halt AR, Stary JM, et al.:
Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices.
Biol Psychiatry.
2002; 52(8): 805–10. PubMed Abstract
| Publisher Full Text
- 15.
Beasley CL, Reynolds GP:
Parvalbumin-immunoreactive neurons are reduced in the prefrontal cortex of schizophrenics.
Schizophr Res.
1997; 24(3): 349–55. PubMed Abstract
| Publisher Full Text
- 16.
Beasley CL, Zhang ZJ, Patten I, et al.:
Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins.
Biol Psychiatry.
2002; 52(7): 708–15. PubMed Abstract
| Publisher Full Text
- 17.
Sakai T, Oshima A, Nozaki Y, et al.:
Changes in density of calcium-binding-protein-immunoreactive GABAergic neurons in prefrontal cortex in schizophrenia and bipolar disorder.
Neuropathology.
2008; 28(2): 143–50. PubMed Abstract
| Publisher Full Text
- 18.
Fung SJ, Webster MJ, Sivagnanasundaram S, et al.:
Expression of interneuron markers in the dorsolateral prefrontal cortex of the developing human and in schizophrenia.
Am J Psychiatry.
2010; 167(12): 1479–88. PubMed Abstract
| Publisher Full Text
- 19.
Selemon LD, Zecevic N:
Schizophrenia: a tale of two critical periods for prefrontal cortical development.
Transl Psychiatry.
2015; 5: e623. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 20.
Hashimoto T, Volk DW, Eggan SM, et al.:
Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia.
J Neurosci.
2003; 23(15): 6315–26. PubMed Abstract
- 21.
Enwright JF, Sanapala S, Foglio A, et al.:
Reduced Labeling of Parvalbumin Neurons and Perineuronal Nets in the Dorsolateral Prefrontal Cortex of Subjects with Schizophrenia.
Neuropsychopharmacology.
2016; 41(9): 2206–14. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 22.
Filice F, Vörckel KJ, Sungur AÖ, et al.:
Reduction in parvalbumin expression not loss of the parvalbumin-expressing GABA interneuron subpopulation in genetic parvalbumin and shank mouse models of autism.
Mol Brain.
2016; 9(10): 10. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 23.
Donato F, Rompani SB, Caroni P:
Parvalbumin-expressing basket-cell network plasticity induced by experience regulates adult learning.
Nature.
2013; 504(7479): 272–6. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 24.
Philpot BD, Lim JH, Brunjes PC:
Activity-dependent regulation of calcium-binding proteins in the developing rat olfactory bulb.
J Comp Neurol.
1997; 387(1): 12–26. PubMed Abstract
| Publisher Full Text
- 25.
Hanno-Iijima Y, Tanaka M, Iijima T:
Activity-Dependent Bidirectional Regulation of GAD Expression in a Homeostatic Fashion Is Mediated by BDNF-Dependent and Independent Pathways.
PLoS One.
2015; 10(8): e0134296. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 26.
Fung SJ, Fillman SG, Webster MJ, et al.:
Schizophrenia and bipolar disorder show both common and distinct changes in cortical interneuron markers.
Schizophr Res.
2014; 155(1–3): 26–30. PubMed Abstract
| Publisher Full Text
- 27.
Klempan TA, Sequeira A, Canetti L, et al.:
Altered expression of genes involved in ATP biosynthesis and GABAergic neurotransmission in the ventral prefrontal cortex of suicides with and without major depression.
Mol Psychiatry.
2009; 14(2): 175–89. PubMed Abstract
| Publisher Full Text
- 28.
Luscher B, Shen Q, Sahir N:
The GABAergic deficit hypothesis of major depressive disorder.
Mol Psychiatry.
2011; 16(4): 383–406. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 29.
Guidotti A, Auta J, Davis JM, et al.:
Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study.
Arch Gen Psychiatry.
2000; 57(11): 1061–9. PubMed Abstract
| Publisher Full Text
- 30.
Fukata Y, Fukata M:
Epilepsy and synaptic proteins.
Curr Opin Neurobiol.
2017; 45(1–8): 1–8. PubMed Abstract
| Publisher Full Text
- 31.
Treiman DM:
GABAergic mechanisms in epilepsy.
Epilepsia.
2001; 42(Suppl 3): 8–12. PubMed Abstract
| Publisher Full Text
- 32.
Symonds C:
Excitation and inhibition in epilepsy.
Proc R Soc Med.
1959; 52(6): 395–402. PubMed Abstract
| Free Full Text
- 33.
Wong M:
Too much inhibition leads to excitation in absence epilepsy.
Epilepsy Curr.
2010; 10(5): 131–2. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 34.
Bolton PF, Carcani-Rathwell I, Hutton J, et al.:
Epilepsy in autism: features and correlates.
Br J Psychiatry.
2011; 198(4): 289–94. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 35.
Tuchman R, Moshé SL, Rapin I:
Convulsing toward the pathophysiology of autism.
Brain Dev.
2009; 31(2): 95–103. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 36.
Jian L, Nagarajan L, de Klerk N, et al.:
Predictors of seizure onset in Rett syndrome.
J Pediatr.
2006; 149(4): 542–7. PubMed Abstract
| Publisher Full Text
- 37.
Amir RE, Van den Veyver IB, Wan M, et al.:
Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2.
Nat Genet.
1999; 23(2): 185–8. PubMed Abstract
| Publisher Full Text
- 38.
Gelisse P, Samuelian JC, Genton P:
Is schizophrenia a risk factor for epilepsy or acute symptomatic seizures?
Epilepsia.
1999; 40(11): 1566–71. PubMed Abstract
| Publisher Full Text
- 39.
Mäkikyrö T, Karvonen JT, Hakko H, et al.:
Comorbidity of hospital-treated psychiatric and physical disorders with special reference to schizophrenia: a 28 year follow-up of the 1966 northern Finland general population birth cohort.
Public Health.
1998; 112(4): 221–8. PubMed Abstract
- 40.
Cascella NG, Schretlen DJ, Sawa A:
Schizophrenia and epilepsy: is there a shared susceptibility?
Neurosci Res.
2009; 63(4): 227–35. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 41.
Reilly C, Atkinson P, Das KB, et al.:
Neurobehavioral comorbidities in children with active epilepsy: a population-based study.
Pediatrics.
2014; 133(6): e1586–93. PubMed Abstract
| Publisher Full Text
- 42.
Williams AE, Giust JM, Kronenberger WG, et al.:
Epilepsy and attention-deficit hyperactivity disorder: links, risks, and challenges.
Neuropsychiatr Dis Treat.
2016; 12: 287–96. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 43.
Buzsáki G, Wang XJ:
Mechanisms of gamma oscillations.
Annu Rev Neurosci.
2012; 35: 203–25. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 44.
Womelsdorf T, Fries P, Mitra PP, et al.:
Gamma-band synchronization in visual cortex predicts speed of change detection.
Nature.
2006; 439(7077): 733–6. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 45.
Tallon-Baudry C, Bertrand O, Peronnet F, et al.:
Induced gamma-band activity during the delay of a visual short-term memory task in humans.
J Neurosci.
1998; 18(11): 4244–54. PubMed Abstract
- 46.
Gonzalez-Burgos G, Lewis DA:
GABA neurons and the mechanisms of network oscillations: implications for understanding cortical dysfunction in schizophrenia.
Schizophr Bull.
2008; 34(5): 944–61. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 47.
Cardin JA, Carlén M, Meletis K, et al.:
Driving fast-spiking cells induces gamma rhythm and controls sensory responses.
Nature.
2009; 459(7247): 663–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 48.
Sohal VS, Zhang F, Yizhar O, et al.:
Parvalbumin neurons and gamma rhythms enhance cortical circuit performance.
Nature.
2009; 459(7247): 698–702. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 49.
Shin YW, O'Donnell BF, Youn S, et al.:
Gamma oscillation in schizophrenia.
Psychiatry Investig.
2011; 8(4): 288–96. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 50.
Chen CM, Stanford AD, Mao X, et al.:
GABA level, gamma oscillation, and working memory performance in schizophrenia.
Neuroimage Clin.
2014; 4: 531–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 51.
Farzan F, Barr MS, Levinson AJ, et al.:
Evidence for gamma inhibition deficits in the dorsolateral prefrontal cortex of patients with schizophrenia.
Brain.
2010; 133(Pt 5): 1505–14. PubMed Abstract
| Publisher Full Text
- 52.
Gordon E, Williams L, Haig AR, et al.:
Symptom profile and “gamma” processing in schizophrenia.
Cogn Neuropsychiatry.
2001; 6(1): 7–19. Publisher Full Text
- 53.
Baldeweg T, Spence S, Hirsch SR, et al.:
Gamma-band electroencephalographic oscillations in a patient with somatic hallucinations.
Lancet.
1998; 352(9128): 620–1. PubMed Abstract
| Publisher Full Text
- 54.
Landau ID, Egger R, Dercksen VJ, et al.:
The Impact of Structural Heterogeneity on Excitation-Inhibition Balance in Cortical Networks.
Neuron.
2016; 92(5): 1106–21. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 55.
Vierling-Claassen D, Siekmeier P, Stufflebeam S, et al.:
Modeling GABA alterations in schizophrenia: a link between impaired inhibition and altered gamma and beta range auditory entrainment.
J Neurophysiol.
2008; 99(5): 2656–71. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 56.
Uhlhaas PJ, Singer W:
Abnormal neural oscillations and synchrony in schizophrenia.
Nat Rev Neurosci.
2010; 11(2): 100–13. PubMed Abstract
| Publisher Full Text
- 57.
Rojas DC, Maharajh K, Teale P, et al.:
Reduced neural synchronization of gamma-band MEG oscillations in first-degree relatives of children with autism.
BMC Psychiatry.
2008; 8: 66. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 58.
Rojas DC, Wilson LB:
γ-band abnormalities as markers of autism spectrum disorders.
Biomark Med.
2014; 8(3): 353–68. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 59.
Lenz D, Krauel K, Schadow J, et al.:
Enhanced gamma-band activity in ADHD patients lacks correlation with memory performance found in healthy children.
Brain Res.
2008; 1235: 117–32. PubMed Abstract
| Publisher Full Text
- 60.
Kamida A, Shimabayashi K, Oguri M, et al.:
EEG Power Spectrum Analysis in Children with ADHD.
Yonago Acta Med.
2016; 59(2): 169–73. PubMed Abstract
| Free Full Text
| Faculty Opinions Recommendation
- 61.
Karch S, Segmiller F, Hantschk I, et al.:
Increased γ oscillations during voluntary selection processes in adult patients with attention deficit/hyperactivity disorder.
J Psychiatr Res.
2012; 46(11): 1515–23. PubMed Abstract
| Publisher Full Text
- 62.
Yordanova J, Banaschewski T, Kolev V, et al.:
Abnormal early stages of task stimulus processing in children with attention-deficit hyperactivity disorder--evidence from event-related gamma oscillations.
Clin Neurophysiol.
2001; 112(6): 1096–108. PubMed Abstract
| Publisher Full Text
- 63.
Özerdem A, Güntekin B, Atagün I, et al.:
Reduced long distance gamma (28–48 Hz) coherence in euthymic patients with bipolar disorder.
J Affect Disord.
2011; 132(3): 325–32. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 64.
Stroganova TA, Butorina AV, Sysoeva OV, et al.:
Altered modulation of gamma oscillation frequency by speed of visual motion in children with autism spectrum disorders.
J Neurodev Disord.
2015; 7(1): 21. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 65.
Petilla Interneuron Nomenclature Group, Ascoli GA, Alonso-Nanclares L, et al.:
Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex.
Nat Rev Neurosci.
2008; 9(7): 557–68. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 66.
DeFelipe J, López-Cruz PL, Benavides-Piccione R, et al.:
New insights into the classification and nomenclature of cortical GABAergic interneurons.
Nat Rev Neurosci.
2013; 14(3): 202–16. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 67.
Kepecs A, Fishell G:
Interneuron cell types are fit to function.
Nature.
2014; 505(7483): 318–26. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 68.
Rudy B, Fishell G, Lee S, et al.:
Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons.
Dev Neurobiol.
2011; 71(1): 45–61. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 69.
Pi H, Hangya B, Kvitsiani D, et al.:
Cortical interneurons that specialize in disinhibitory control.
Nature.
2013; 503(7477): 521–4. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 70.
Lee S, Kruglikov I, Huang ZJ, et al.:
A disinhibitory circuit mediates motor integration in the somatosensory cortex.
Nat Neurosci.
2013; 16(11): 1662–70. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 71.
Fu Y, Tucciarone JM, Espinosa JS, et al.:
A cortical circuit for gain control by behavioral state.
Cell.
2014; 156(6): 1139–52. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 72.
Pfeffer CK, Xue M, He M, et al.:
Inhibition of inhibition in visual cortex: the logic of connections between molecularly distinct interneurons.
Nat Neurosci.
2013; 16(8): 1068–76. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 73.
Jackson J, Ayzenshtat I, Karnani MM, et al.:
VIP+ interneurons control neocortical activity across brain states.
J Neurophysiol.
2016; 115(6): 3008–17. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 74.
Batista-Brito R, Vinck M, Ferguson KA, et al.:
Developmental Dysfunction of VIP Interneurons Impairs Cortical Circuits.
Neuron.
2017; 95(4): 884–895.e9. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 75.
Garcia-Junco-Clemente P, Ikrar T, Tring E, et al.:
An inhibitory pull-push circuit in frontal cortex.
Nat Neurosci.
2017; 20(3): 389–92. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 76.
Vacic V, McCarthy S, Malhotra D, et al.:
Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia.
Nature.
2011; 471(7339): 499–503. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 77.
Chiu CQ, Lur G, Morse TM, et al.:
Compartmentalization of GABAergic inhibition by dendritic spines.
Science.
2013; 340(6133): 759–62. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 78.
Gentet LJ, Kremer Y, Taniguchi H, et al.:
Unique functional properties of somatostatin-expressing GABAergic neurons in mouse barrel cortex.
Nat Neurosci.
2012; 15(4): 607–12. PubMed Abstract
| Publisher Full Text
- 79.
Scheyltjens I, Arckens L:
The Current Status of Somatostatin-Interneurons in Inhibitory Control of Brain Function and Plasticity.
Neural Plast.
2016; 2016: 8723623. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 80.
Cichon J, Gan W:
Branch-specific dendritic Ca2+ spikes cause persistent synaptic plasticity.
Nature.
2015; 520(7546): 180–5. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 81.
Fuchs T, Jefferson SJ, Hooper A, et al.:
Disinhibition of somatostatin-positive GABAergic interneurons results in an anxiolytic and antidepressant-like brain state.
Mol Psychiatry.
2017; 22(6): 920–30. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 82.
Lin L, Sibille E:
Reduced brain somatostatin in mood disorders: a common pathophysiological substrate and drug target?
Front Pharmacol.
2013; 4: 110. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 83.
Veit J, Hakim R, Jadi MP, et al.:
Cortical gamma band synchronization through somatostatin interneurons.
Nat Neurosci.
2017; 20(7): 951–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 84.
Bezaire MJ, Soltesz I:
Quantitative assessment of CA1 local circuits: knowledge base for interneuron-pyramidal cell connectivity.
Hippocampus.
2013; 23(9): 751–85. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 85.
Hu H, Gan J, Jonas P:
Interneurons. Fast-spiking, parvalbumin+ GABAergic interneurons: from cellular design to microcircuit function.
Science.
2014; 345(6196): 1255263. PubMed Abstract
| Publisher Full Text
- 86.
Nelson SB, Valakh V:
Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders.
Neuron.
2015; 87(4): 684–98. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 87.
Klausberger T, Somogyi P:
Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations.
Science.
2008; 321(5885): 53–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 88.
Tricoire L, Pelkey KA, Erkkila BE, et al.:
A blueprint for the spatiotemporal origins of mouse hippocampal interneuron diversity.
J Neurosci.
2011; 31(30): 10948–70. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 89.
Pouille F, Scanziani M:
Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition.
Science.
2001; 293(5532): 1159–63. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 90.
Hu H, Jonas P:
A supercritical density of Na+ channels ensures fast signaling in GABAergic interneuron axons.
Nat Neurosci.
2014; 17(5): 686–93. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 91.
Ogiwara I, Miyamoto H, Morita N, et al.:
Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation.
J Neurosci.
2007; 27(22): 5903–14. PubMed Abstract
| Publisher Full Text
- 92.
Zaitsev AV, Povysheva NV, Lewis DA, et al.:
P/Q-type, but not N-type, calcium channels mediate GABA release from fast-spiking interneurons to pyramidal cells in rat prefrontal cortex.
J Neurophysiol.
2007; 97(5): 3567–73. PubMed Abstract
| Publisher Full Text
- 93.
Bucurenciu I, Kulik A, Schwaller B, et al.:
Nanodomain coupling between Ca2+ channels and Ca2+ sensors promotes fast and efficient transmitter release at a cortical GABAergic synapse.
Neuron.
2008; 57(4): 536–45. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 94.
Baude A, Bleasdale C, Dalezios Y, et al.:
Immunoreactivity for the GABAA receptor alpha1 subunit, somatostatin and Connexin36 distinguishes axoaxonic, basket, and bistratified interneurons of the rat hippocampus.
Cereb Cortex.
2007; 17(9): 2094–107. PubMed Abstract
| Publisher Full Text
- 95.
Palay SL, Sotelo C, Peters A, et al.:
The axon hillock and the initial segment.
J Cell Biol.
1968; 38(1): 193–201. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 96.
Somogyi P:
A specific 'axo-axonal' interneuron in the visual cortex of the rat.
Brain Res.
1977; 136(2): 345–50. PubMed Abstract
| Publisher Full Text
- 97.
Inan M, Blázquez-Llorca L, Merchán-Pérez A, et al.:
Dense and overlapping innervation of pyramidal neurons by chandelier cells.
J Neurosci.
2013; 33(5): 1907–14. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 98.
Wang Y, Zhang P, Wyskiel DR:
Chandelier Cells in Functional and Dysfunctional Neural Circuits.
Front Neural Circuits.
2016; 10: 33. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 99.
Inda MC, Defelipe J, Muñoz A:
The distribution of chandelier cell axon terminals that express the GABA plasma membrane transporter GAT-1 in the human neocortex.
Cereb Cortex.
2007; 17(9): 2060–71. PubMed Abstract
| Publisher Full Text
- 100.
Cruz DA, Eggan SM, Lewis DA:
Postnatal development of pre- and postsynaptic GABA markers at chandelier cell connections with pyramidal neurons in monkey prefrontal cortex.
J Comp Neurol.
2003; 465(3): 385–400. PubMed Abstract
| Publisher Full Text
- 101.
Nusser Z, Sieghart W, Benke D, et al.:
Differential synaptic localization of two major gamma-aminobutyric acid type A receptor alpha subunits on hippocampal pyramidal cells.
Proc Natl Acad Sci U S A.
1996; 93(21): 11939–44. PubMed Abstract
| Free Full Text
- 102.
Nyíri G, Freund TF, Somogyi P:
Input-dependent synaptic targeting of alpha2-subunit-containing GABAA receptors in synapses of hippocampal pyramidal cells of the rat.
Eur J Neurosci.
2001; 13(3): 428–42. PubMed Abstract
| Publisher Full Text
- 103.
Chiu CS, Jensen K, Sokolova I, et al.:
Number, density, and surface/cytoplasmic distribution of GABA transporters at presynaptic structures of knock-in mice carrying GABA transporter subtype 1-green fluorescent protein fusions.
J Neurosci.
2002; 22(23): 10251–66. PubMed Abstract
- 104.
Zhu Y, Stornetta RL, Zhu JJ:
Chandelier cells control excessive cortical excitation: characteristics of whisker-evoked synaptic responses of layer 2/3 nonpyramidal and pyramidal neurons.
J Neurosci.
2004; 24(22): 5101–8. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 105.
Ribak CE:
Axon terminals of GABAergic chandelier cells are lost at epileptic foci.
Brain Res.
1985; 326(2): 251–60. PubMed Abstract
| Publisher Full Text
- 106.
Defelipe J:
Chandelier cells and epilepsy.
Brain.
1999; 122(Pt 10): 1807–22. PubMed Abstract
| Publisher Full Text
- 107.
Dinocourt C, Petanjek Z, Freund TF, et al.:
Loss of interneurons innervating pyramidal cell dendrites and axon initial segments in the CA1 region of the hippocampus following pilocarpine-induced seizures.
J Comp Neurol.
2003; 459(4): 407–25. PubMed Abstract
| Publisher Full Text
- 108.
Woodruff A, Xu Q, Anderson SA, et al.:
Depolarizing effect of neocortical chandelier neurons.
Front Neural Circuits.
2009; 3: 15. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 109.
Szabadics J, Varga C, Molnár G, et al.:
Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits.
Science.
2006; 311(5758): 233–5. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 110.
Wang X, Hooks BM, Sun QQ:
Thorough GABAergic innervation of the entire axon initial segment revealed by an optogenetic 'laserspritzer'.
J Physiol.
2014; 592(19): 4257–76. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 111.
Woodruff AR, McGarry LM, Vogels TP, et al.:
State-dependent function of neocortical chandelier cells.
J Neurosci.
2011; 31(49): 17872–86. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 112.
Massi L, Lagler M, Hartwich K, et al.:
Temporal dynamics of parvalbumin-expressing axo-axonic and basket cells in the rat medial prefrontal cortex in vivo.
J Neurosci.
2012; 32(46): 16496–502. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 113.
Viney TJ, Lasztoczi B, Katona L, et al.:
Network state-dependent inhibition of identified hippocampal CA3 axo-axonic cells in vivo.
Nat Neurosci.
2013; 16(12): 1802–11. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 114.
Rocco BR, DeDionisio AM, Lewis DA, et al.:
Alterations in a Unique Class of Cortical Chandelier Cell Axon Cartridges in Schizophrenia.
Biol Psychiatry.
2017; 82(1): 40–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 115.
Pierri JN, Chaudry AS, Woo TU, et al.:
Alterations in chandelier neuron axon terminals in the prefrontal cortex of schizophrenic subjects.
Am J Psychiatry.
1999; 156(11): 1709–19. PubMed Abstract
- 116.
Schleimer SB, Hinton T, Dixon G, et al.:
GABA transporters GAT-1 and GAT-3 in the human dorsolateral prefrontal cortex in schizophrenia.
Neuropsychobiology.
2004; 50(3): 226–30. PubMed Abstract
| Publisher Full Text
- 117.
Volk D, Austin M, Pierri J, et al.:
GABA transporter-1 mRNA in the prefrontal cortex in schizophrenia: decreased expression in a subset of neurons.
Am J Psychiatry.
2001; 158(2): 256–65. PubMed Abstract
| Publisher Full Text
- 118.
Volk DW, Pierri JN, Fritschy JM, et al.:
Reciprocal alterations in pre- and postsynaptic inhibitory markers at chandelier cell inputs to pyramidal neurons in schizophrenia.
Cereb Cortex.
2002; 12(10): 1063–70. PubMed Abstract
- 119.
Howard A, Tamas G, Soltesz I:
Lighting the chandelier: new vistas for axo-axonic cells.
Trends Neurosci.
2005; 28(6): 310–6. PubMed Abstract
| Publisher Full Text
- 120.
Del Pino I, García-Frigola C, Dehorter N, et al.:
Erbb4 deletion from fast-spiking interneurons causes schizophrenia-like phenotypes.
Neuron.
2013; 79(6): 1152–68. PubMed Abstract
| Publisher Full Text
- 121.
Ting AK, Chen Y, Wen L, et al.:
Neuregulin 1 promotes excitatory synapse development and function in GABAergic interneurons.
J Neurosci.
2011; 31(1): 15–25. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 122.
Gibson JR, Bartley AF, Hays SA, et al.:
Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome.
J Neurophysiol.
2008; 100(5): 2615–26. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 123.
Seshadri S, Faust T, Ishizuka K, et al.:
Interneuronal DISC1 regulates NRG1-ErbB4 signalling and excitatory-inhibitory synapse formation in the mature cortex.
Nat Commun.
2015; 6: 10118. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 124.
Polepalli JS, Wu H, Goswami D, et al.:
Modulation of excitation on parvalbumin interneurons by neuroligin-3 regulates the hippocampal network.
Nat Neurosci.
2017; 20(2): 219–29. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 125.
He LJ, Liu N, Cheng TL, et al.:
Conditional deletion of Mecp2 in parvalbumin-expressing GABAergic cells results in the absence of critical period plasticity.
Nat Commun.
2014; 5(1): 5036. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 126.
Ji Y, Yang F, Papaleo F, et al.:
Role of dysbindin in dopamine receptor trafficking and cortical GABA function.
Proc Natl Acad Sci U S A.
2009; 106(46): 19593–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 127.
Yu FH, Mantegazza M, Westenbroek RE, et al.:
Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy.
Nat Neurosci.
2006; 9(9): 1142–9. PubMed Abstract
| Publisher Full Text
- 128.
Fazzari P, Paternain AV, Valiente M, et al.:
Control of cortical GABA circuitry development by Nrg1 and ErbB4 signalling.
Nature.
2010; 464(7293): 1376–80. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 129.
Bateup HS, Johnson CA, Denefrio CL, et al.:
Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis.
Neuron.
2013; 78(3): 510–22. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 130.
Wallace ML, Burette AC, Weinberg RJ, et al.:
Maternal loss of Ube3a produces an excitatory/inhibitory imbalance through neuron type-specific synaptic defects.
Neuron.
2012; 74(5): 793–800. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 131.
Lee J, Chung C, Ha S, et al.:
Shank3-mutant mice lacking exon 9 show altered excitation/inhibition balance, enhanced rearing, and spatial memory deficit.
Front Cell Neurosci.
2015; 9: 94. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 132.
Won H, Mah W, Kim E, et al.:
GIT1 is associated with ADHD in humans and ADHD-like behaviors in mice.
Nat Med.
2011; 17(5): 566–72. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 133.
Rivero O, Selten MM, Sich S, et al.:
Cadherin-13, a risk gene for ADHD and comorbid disorders, impacts GABAergic function in hippocampus and cognition.
Transl Psychiatry.
2015; 5: e655. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 134.
Gibson JR, Huber KM, Südhof TC:
Neuroligin-2 deletion selectively decreases inhibitory synaptic transmission originating from fast-spiking but not from somatostatin-positive interneurons.
J Neurosci.
2009; 29(44): 13883–97. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 135.
Poulopoulos A, Aramuni G, Meyer G, et al.:
Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin.
Neuron.
2009; 63(5): 628–42. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 136.
Földy C, Malenka RC, Südhof TC:
Autism-associated neuroligin-3 mutations commonly disrupt tonic endocannabinoid signaling.
Neuron.
2013; 78(3): 498–509. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 137.
Anderson GR, Galfin T, Xu W, et al.:
Candidate autism gene screen identifies critical role for cell-adhesion molecule CASPR2 in dendritic arborization and spine development.
Proc Natl Acad Sci U S A.
2012; 109(44): 18120–5. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 138.
Xue M, Atallah BV, Scanziani M:
Equalizing excitation-inhibition ratios across visual cortical neurons.
Nature.
2014; 511(7511): 596–600. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 139.
Tamás G, Buhl EH, Lörincz A, et al.:
Proximally targeted GABAergic synapses and gap junctions synchronize cortical interneurons.
Nat Neurosci.
2000; 3(4): 366–71. PubMed Abstract
| Publisher Full Text
- 140.
Taniguchi H, Lu J, Huang ZJ:
The spatial and temporal origin of chandelier cells in mouse neocortex.
Science.
2013; 339(6115): 70–4. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 141.
Bartos M, Vida I, Jonas P:
Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks.
Nat Rev Neurosci.
2007; 8(1): 45–56. PubMed Abstract
| Publisher Full Text
- 142.
Traub RD, Kopell N, Bibbig A, et al.:
Gap junctions between interneuron dendrites can enhance synchrony of gamma oscillations in distributed networks.
J Neurosci.
2001; 21(23): 9478–86. PubMed Abstract
| Faculty Opinions Recommendation
- 143.
Mullins C, Fishell G, Tsien RW:
Unifying Views of Autism Spectrum Disorders: A Consideration of Autoregulatory Feedback Loops.
Neuron.
2016; 89(6): 1131–56. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 144.
Turrigiano GG, Leslie KR, Desai NS, et al.:
Activity-dependent scaling of quantal amplitude in neocortical neurons.
Nature.
1998; 391(6670): 892–6. PubMed Abstract
| Publisher Full Text
- 145.
Kilman V, van Rossum MC, Turrigiano GG:
Activity deprivation reduces miniature IPSC amplitude by decreasing the number of postsynaptic GABA(A) receptors clustered at neocortical synapses.
J Neurosci.
2002; 22(4): 1328–37. PubMed Abstract
| Faculty Opinions Recommendation
- 146.
Desai NS, Rutherford LC, Turrigiano GG:
Plasticity in the intrinsic excitability of cortical pyramidal neurons.
Nat Neurosci.
1999; 2(6): 515–20. PubMed Abstract
| Publisher Full Text
- 147.
Bartley AF, Huang ZJ, Huber KM, et al.:
Differential activity-dependent, homeostatic plasticity of two neocortical inhibitory circuits.
J Neurophysiol.
2008; 100(4): 1983–94. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 148.
Echegoyen J, Neu A, Graber KD, et al.:
Homeostatic plasticity studied using in vivo hippocampal activity-blockade: synaptic scaling, intrinsic plasticity and age-dependence.
PLoS One.
2007; 2(8): e700. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 149.
Norton N, Moskvina V, Morris DW, et al.:
Evidence that interaction between neuregulin 1 and its receptor erbB4 increases susceptibility to schizophrenia.
Am J Med Genet B Neuropsychiatr Genet.
2006; 141B(1): 96–101. PubMed Abstract
| Publisher Full Text
- 150.
Silberberg G, Darvasi A, Pinkas-Kramarski R, et al.:
The involvement of ErbB4 with schizophrenia: association and expression studies.
Am J Med Genet B Neuropsychiatr Genet.
2006; 141B(2): 142–8. PubMed Abstract
| Publisher Full Text
- 151.
Vullhorst D, Neddens J, Karavanova I, et al.:
Selective expression of ErbB4 in interneurons, but not pyramidal cells, of the rodent hippocampus.
J Neurosci.
2009; 29(39): 12255–64. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 152.
Woo RS, Li XM, Tao Y, et al.:
Neuregulin-1 enhances depolarization-induced GABA release.
Neuron.
2007; 54(4): 599–610. PubMed Abstract
| Publisher Full Text
- 153.
Stefansson H, Sigurdsson E, Steinthorsdottir V, et al.:
Neuregulin 1 and susceptibility to schizophrenia.
Am J Hum Genet.
2002; 71(4): 877–92. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 154.
Georgieva L, Dimitrova A, Ivanov D, et al.:
Support for neuregulin 1 as a susceptibility gene for bipolar disorder and schizophrenia.
Biol Psychiatry.
2008; 64(5): 419–27. PubMed Abstract
| Publisher Full Text
- 155.
Prata DP, Breen G, Osborne S, et al.:
An association study of the neuregulin 1 gene, bipolar affective disorder and psychosis.
Psychiatr Genet.
2009; 19(3): 113–6. PubMed Abstract
| Publisher Full Text
- 156.
Verkerk AJ, Pieretti M, Sutcliffe JS, et al.:
Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome.
Cell.
1991; 65(5): 905–14. PubMed Abstract
| Publisher Full Text
- 157.
Kaufmann WE, Cortell R, Kau AS, et al.:
Autism spectrum disorder in fragile X syndrome: communication, social interaction, and specific behaviors.
Am J Med Genet A.
2004; 129A(3): 225–34. PubMed Abstract
| Publisher Full Text
- 158.
Bear MF, Huber KM, Warren ST:
The mGluR theory of fragile X mental retardation.
Trends Neurosci.
2004; 27(7): 370–7. PubMed Abstract
| Publisher Full Text
- 159.
Paluszkiewicz SM, Martin BS, Huntsman MM:
Fragile X syndrome: the GABAergic system and circuit dysfunction.
Dev Neurosci.
2011; 33(5): 349–64. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 160.
Cea-Del Rio CA, Huntsman MM:
The contribution of inhibitory interneurons to circuit dysfunction in Fragile X Syndrome.
Front Cell Neurosci.
2014; 8: 245. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 161.
D'Hulst C, De Geest N, Reeve SP, et al.:
Decreased expression of the GABAA receptor in fragile X syndrome.
Brain Res.
2006; 1121(1): 238–45. PubMed Abstract
| Publisher Full Text
- 162.
Selby L, Zhang C, Sun QQ:
Major defects in neocortical GABAergic inhibitory circuits in mice lacking the fragile X mental retardation protein.
Neurosci Lett.
2007; 412(3): 227–32. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 163.
Gatto CL, Broadie K:
Genetic controls balancing excitatory and inhibitory synaptogenesis in neurodevelopmental disorder models.
Front Synaptic Neurosci.
2010; 2: 4. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 164.
Krishnan K, Wang BS, Lu J, et al.:
MeCP2 regulates the timing of critical period plasticity that shapes functional connectivity in primary visual cortex.
Proc Natl Acad Sci U S A.
2015; 112(34): E4782–91. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 165.
Hensch TK:
Critical period plasticity in local cortical circuits.
Nat Rev Neurosci.
2005; 6(11): 877–88. PubMed Abstract
| Publisher Full Text
- 166.
Marín O:
Developmental timing and critical windows for the treatment of psychiatric disorders.
Nat Med.
2016; 22(11): 1229–38. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 167.
Chao HT, Chen H, Samaco RC, et al.:
Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes.
Nature.
2010; 468(7321): 263–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 168.
Ito-Ishida A, Ure K, Chen H, et al.:
Loss of MeCP2 in Parvalbumin-and Somatostatin-Expressing Neurons in Mice Leads to Distinct Rett Syndrome-like Phenotypes.
Neuron.
2015; 88(4): 651–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 169.
Banerjee A, Rikhye RV, Breton-Provencher V, et al.:
Jointly reduced inhibition and excitation underlies circuit-wide changes in cortical processing in Rett syndrome.
Proc Natl Acad Sci U S A.
2016; 113(46): E7287–E7296. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 170.
Dehorter N, Marichal N, Marín O, et al.:
Tuning neural circuits by turning the interneuron knob.
Curr Opin Neurobiol.
2017; 42: 144–51. PubMed Abstract
| Publisher Full Text
- 171.
Beck H, Yaari Y:
Plasticity of intrinsic neuronal properties in CNS disorders.
Nat Rev Neurosci.
2008; 9(5): 357–69. PubMed Abstract
| Publisher Full Text
- 172.
Straub RE, Jiang Y, MacLean CJ, et al.:
Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia.
Am J Hum Genet.
2002; 71(2): 337–48. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 173.
Carlson GC, Talbot K, Halene TB, et al.:
Dysbindin-1 mutant mice implicate reduced fast-phasic inhibition as a final common disease mechanism in schizophrenia.
Proc Natl Acad Sci U S A.
2011; 108(43): E962–70. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 174.
Larimore J, Zlatic SA, Arnold M, et al.:
Dysbindin Deficiency Modifies the Expression of GABA Neuron and Ion Permeation Transcripts in the Developing Hippocampus.
Front Genet.
2017; 8: 28. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 175.
Li BM, Liu XR, Yi YH, et al.:
Autism in Dravet syndrome: prevalence, features, and relationship to the clinical characteristics of epilepsy and mental retardation.
Epilepsy Behav.
2011; 21(3): 291–5. PubMed Abstract
| Publisher Full Text
- 176.
O'Roak BJ, Deriziotis P, Lee C, et al.:
Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations.
Nat Genet.
2011; 43(6): 585–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 177.
Van Wart A, Trimmer JS, Matthews G:
Polarized distribution of ion channels within microdomains of the axon initial segment.
J Comp Neurol.
2007; 500(2): 339–52. PubMed Abstract
| Publisher Full Text
- 178.
Kole MH, Stuart GJ:
Signal processing in the axon initial segment.
Neuron.
2012; 73(2): 235–47. PubMed Abstract
| Publisher Full Text
- 179.
Cheah CS, Yu FH, Westenbroek RE, et al.:
Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome.
Proc Natl Acad Sci U S A.
2012; 109(36): 14646–51. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 180.
Dutton SB, Makinson CD, Papale LA, et al.:
Preferential inactivation of Scn1a in parvalbumin interneurons increases seizure susceptibility.
Neurobiol Dis.
2013; 49: 211–20. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 181.
Han S, Tai C, Westenbroek RE, et al.:
Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission.
Nature.
2012; 489(7416): 385–90. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 182.
Stedehouder J, Kushner SA:
Myelination of parvalbumin interneurons: a parsimonious locus of pathophysiological convergence in schizophrenia.
Mol Psychiatry.
2017; 22(1): 4–12. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 183.
White T, Magnotta VA, Bockholt HJ, et al.:
Global white matter abnormalities in schizophrenia: a multisite diffusion tensor imaging study.
Schizophr Bull.
2011; 37(1): 222–32. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 184.
Uranova NA, Vostrikov VM, Orlovskaya DD, et al.:
Oligodendroglial density in the prefrontal cortex in schizophrenia and mood disorders: a study from the Stanley Neuropathology Consortium.
Schizophr Res.
2004; 67(2–3): 269–75. PubMed Abstract
| Publisher Full Text
- 185.
Hakak Y, Walker JR, Li C, et al.:
Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia.
Proc Natl Acad Sci U S A.
2001; 98(8): 4746–51. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 186.
Katsumaru H, Kosaka T, Heizmann CW, et al.:
Immunocytochemical study of GABAergic neurons containing the calcium-binding protein parvalbumin in the rat hippocampus.
Exp Brain Res.
1988; 72(2): 347–62. PubMed Abstract
| Publisher Full Text
- 187.
Micheva KD, Wolman D, Mensh BD, et al.:
A large fraction of neocortical myelin ensheathes axons of local inhibitory neurons.
eLife.
2016; 5: pii: e15784. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 188.
Chung K, Wallace J, Kim SY, et al.:
Structural and molecular interrogation of intact biological systems.
Nature.
2013; 497(7449): 332–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 189.
Nave KA, Werner HB:
Myelination of the nervous system: mechanisms and functions.
Annu Rev Cell Dev Biol.
2014; 30: 503–33. PubMed Abstract
| Publisher Full Text
- 190.
Lewis DA:
The chandelier neuron in schizophrenia.
Dev Neurobiol.
2011; 71(1): 118–27. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 191.
Del Pino I, Brotons-Mas JR, Marques-Smith A, et al.:
Abnormal wiring of CCK+ basket cells disrupts spatial information coding.
Nat Neurosci.
2017; 20(6): 784–92. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 192.
Prather P, de Vries PJ:
Behavioral and cognitive aspects of tuberous sclerosis complex.
J Child Neurol.
2004; 19(9): 666–74. PubMed Abstract
| Publisher Full Text
- 193.
Jones AC, Shyamsundar MM, Thomas MW, et al.:
Comprehensive mutation analysis of TSC1 and TSC2-and phenotypic correlations in 150 families with tuberous sclerosis.
Am J Hum Genet.
1999; 64(5): 1305–15. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 194.
Rougeulle C, Glatt H, Lalande M:
The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain.
Nat Genet.
1997; 17(1): 14–5. PubMed Abstract
| Publisher Full Text
- 195.
Williams CA, Beaudet AL, Clayton-Smith J, et al.:
Angelman syndrome 2005: updated consensus for diagnostic criteria.
Am J Med Genet A.
2006; 140(5): 413–8. PubMed Abstract
| Publisher Full Text
- 196.
Thibert RL, Conant KD, Braun EK, et al.:
Epilepsy in Angelman syndrome: a questionnaire-based assessment of the natural history and current treatment options.
Epilepsia.
2009; 50(11): 2369–76. PubMed Abstract
| Publisher Full Text
- 197.
Jiang YH, Armstrong D, Albrecht U, et al.:
Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation.
Neuron.
1998; 21(4): 799–811. PubMed Abstract
| Publisher Full Text
- 198.
Yashiro K, Riday TT, Condon KH, et al.:
Ube3a is required for experience-dependent maturation of the neocortex.
Nat Neurosci.
2009; 12(6): 777–83. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 199.
Judson MC, Wallace ML, Sidorov MS, et al.:
GABAergic Neuron-Specific Loss of Ube3a Causes Angelman Syndrome-Like EEG Abnormalities and Enhances Seizure Susceptibility.
Neuron.
2016; 90(1): 56–69. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 200.
Wallace ML, van Woerden GM, Elgersma Y, et al.:
Ube3a loss increases excitability and blunts orientation tuning in the visual cortex of Angelman syndrome model mice.
J Neurophysiol.
2017; 118(1): 634–46. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 201.
Atallah BV, Bruns W, Carandini M, et al.:
Parvalbumin-expressing interneurons linearly transform cortical responses to visual stimuli.
Neuron.
2012; 73(1): 159–70. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 202.
Wilson NR, Runyan CA, Wang FL, et al.:
Division and subtraction by distinct cortical inhibitory networks in vivo.
Nature.
2012; 488(7411): 343–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 203.
Leblond CS, Nava C, Polge A, et al.:
Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: a gradient of severity in cognitive impairments.
PLoS Genet.
2014; 10(9): e1004580. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 204.
Monteiro P, Feng G:
SHANK proteins: roles at the synapse and in autism spectrum disorder.
Nat Rev Neurosci.
2017; 18(3): 147–57. PubMed Abstract
| Publisher Full Text
- 205.
Naisbitt S, Kim E, Tu JC, et al.:
Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin.
Neuron.
1999; 23(3): 569–82. PubMed Abstract
| Publisher Full Text
- 206.
Tao-Cheng J, Yang Y, Reese TS, et al.:
Differential distribution of Shank and GKAP at the postsynaptic density.
PLoS One.
2015; 10(3): e0118750. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 207.
Peça J, Feliciano C, Ting JT, et al.:
Shank3 mutant mice display autistic-like behaviours and striatal dysfunction.
Nature.
2011; 472(7344): 437–42. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 208.
Bozdagi O, Sakurai T, Papapetrou D, et al.:
Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication.
Mol Autism.
2010; 1(1): 15. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 209.
Wang X, McCoy PA, Rodriguiz RM, et al.:
Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3.
Hum Mol Genet.
2011; 20(15): 3093–108. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 210.
Südhof TC:
Neuroligins and neurexins link synaptic function to cognitive disease.
Nature.
2008; 455(7215): 903–11. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 211.
Chih B, Engelman H, Scheiffele P:
Control of excitatory and inhibitory synapse formation by neuroligins.
Science.
2005; 307(5713): 1324–8. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 212.
Song JY, Ichtchenko K, Südhof TC, et al.:
Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses.
Proc Natl Acad Sci U S A.
1999; 96(3): 1100–5. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 213.
Varoqueaux F, Jamain S, Brose N:
Neuroligin 2 is exclusively localized to inhibitory synapses.
Eur J Cell Biol.
2004; 83(9): 449–56. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 214.
Budreck EC, Scheiffele P:
Neuroligin-3 is a neuronal adhesion protein at GABAergic and glutamatergic synapses.
Eur J Neurosci.
2007; 26(7): 1738–48. PubMed Abstract
| Publisher Full Text
- 215.
Hoon M, Soykan T, Falkenburger B, et al.:
Neuroligin-4 is localized to glycinergic postsynapses and regulates inhibition in the retina.
Proc Natl Acad Sci U S A.
2011; 108(7): 3053–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 216.
Jamain S, Quach H, Betancur C, et al.:
Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism.
Nat Genet.
2003; 34(1): 27–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 217.
Tabuchi K, Blundell J, Etherton MR, et al.:
A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice.
Science.
2007; 318(5847): 71–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 218.
Hammer M, Krueger-Burg D, Tuffy LP, et al.:
Perturbed Hippocampal Synaptic Inhibition and γ-Oscillations in a Neuroligin-4 Knockout Mouse Model of Autism.
Cell Rep.
2015; 13(3): 516–23. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 219.
Parente DJ, Garriga C, Baskin B, et al.:
Neuroligin 2 nonsense variant associated with anxiety, autism, intellectual disability, hyperphagia, and obesity.
Am J Med Genet A.
2017; 173(1): 213–6. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 220.
Sun C, Cheng MC, Qin R, et al.:
Identification and functional characterization of rare mutations of the neuroligin-2 gene (NLGN2) associated with schizophrenia.
Hum Mol Genet.
2011; 20(15): 3042–51. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 221.
Kim HG, Kishikawa S, Higgins AW, et al.:
Disruption of neurexin 1 associated with autism spectrum disorder.
Am J Hum Genet.
2008; 82(1): 199–207. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 222.
Kirov G, Gumus D, Chen W, et al.:
Comparative genome hybridization suggests a role for NRXN1 and APBA2 in schizophrenia.
Hum Mol Genet.
2008; 17(3): 458–65. PubMed Abstract
| Publisher Full Text
- 223.
Kirov G, Rujescu D, Ingason A, et al.:
Neurexin 1 (NRXN1) deletions in schizophrenia.
Schizophr Bull.
2009; 35(5): 851–4. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 224.
Etherton MR, Blaiss CA, Powell CM, et al.:
Mouse neurexin-1alpha deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments.
Proc Natl Acad Sci U S A.
2009; 106(42): 17998–8003. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 225.
Rabaneda LG, Robles-Lanuza E, Nieto-González JL, et al.:
Neurexin dysfunction in adult neurons results in autistic-like behavior in mice.
Cell Rep.
2014; 8(2): 338–46. PubMed Abstract
| Publisher Full Text
- 226.
de la Peña JB, Dela Peña IJ, Custodio RJ, et al.:
Exploring the Validity of Proposed Transgenic Animal Models of Attention-Deficit Hyperactivity Disorder (ADHD).
Mol Neurobiol.
2017; 1–16. PubMed Abstract
| Publisher Full Text
- 227.
Salatino-Oliveira A, Genro JP, Chazan R, et al.:
Association study of GIT1 gene with attention-deficit hyperactivity disorder in Brazilian children and adolescents.
Genes Brain Behav.
2012; 11(7): 864–8. PubMed Abstract
| Publisher Full Text
- 228.
Klein M, van der Voet M, Harich B, et al.:
Converging evidence does not support GIT1 as an ADHD risk gene.
Am J Med Genet B Neuropsychiatr Genet.
2015; 168(6): 492–507. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 229.
Smith KR, Davenport EC, Wei J, et al.:
GIT1 and βPIX are essential for GABAA receptor synaptic stability and inhibitory neurotransmission.
Cell Rep.
2014; 9(1): 298–310. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 230.
Arias-Vásquez A, Altink ME, Rommelse NN, et al.:
CDH13 is associated with working memory performance in attention deficit/hyperactivity disorder.
Genes Brain Behav.
2011; 10(8): 844–51. PubMed Abstract
| Publisher Full Text
- 231.
Zhou K, Dempfle A, Arcos-Burgos M, et al.:
Meta-analysis of genome-wide linkage scans of attention deficit hyperactivity disorder.
Am J Med Genet B Neuropsychiatr Genet.
2008; 147B(8): 1392–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 232.
Rivero O, Sich S, Popp S, et al.:
Impact of the ADHD-susceptibility gene CDH13 on development and function of brain networks.
Eur Neuropsychopharmacol.
2013; 23(6): 492–507. PubMed Abstract
| Publisher Full Text
- 233.
Killen AC, Barber M, Paulin JJW, et al.:
Protective role of Cadherin 13 in interneuron development.
Brain Struct Funct.
2017; 222(8): 3567–3585. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 234.
Benarroch EE:
GABAB receptors: structure, functions, and clinical implications.
Neurology.
2012; 78(8): 578–84. PubMed Abstract
| Publisher Full Text
- 235.
Lüscher C, Jan LY, Stoffel M, et al.:
G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons.
Neuron.
1997; 19(3): 687–95. PubMed Abstract
| Publisher Full Text
- 236.
Ikeda SR:
Voltage-dependent modulation of N-type calcium channels by G-protein beta gamma subunits.
Nature.
1996; 380(6571): 255–8. PubMed Abstract
| Publisher Full Text
- 237.
Mintz IM, Bean BP:
GABAB receptor inhibition of P-type Ca2+ channels in central neurons.
Neuron.
1993; 10(5): 889–98. PubMed Abstract
| Publisher Full Text
- 238.
Mizukami K, Sasaki M, Ishikawa M, et al.:
Immunohistochemical localization of gamma-aminobutyric acidB receptor in the hippocampus of subjects with schizophrenia.
Neurosci Lett.
2000; 283(2): 101–4. PubMed Abstract
| Publisher Full Text
- 239.
Mizukami K, Ishikawa M, Hidaka S, et al.:
Immunohistochemical localization of GABAB receptor in the entorhinal cortex and inferior temporal cortex of schizophrenic brain.
Prog Neuropsychopharmacol Biol Psychiatry.
2002; 26(2): 393–6. PubMed Abstract
| Publisher Full Text
- 240.
Ishikawa M, Mizukami K, Iwakiri M, et al.:
Immunohistochemical and immunoblot analysis of gamma-aminobutyric acid B receptor in the prefrontal cortex of subjects with schizophrenia and bipolar disorder.
Neurosci Lett.
2005; 383(3): 272–7. PubMed Abstract
| Publisher Full Text
- 241.
Fatemi SH, Folsom TD, Thuras PD:
Deficits in GABAB receptor system in schizophrenia and mood disorders: a postmortem study.
Schizophr Res.
2011; 128(1–3): 37–43. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 242.
Selten MM, Meyer F, Ba W, et al.:
Increased GABAB receptor signaling in a rat model for schizophrenia.
Sci Rep.
2016; 6: 34240. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 243.
Wierońska JM, Kusek M, Tokarski K, et al.:
The GABAB receptor agonist CGP44532 and the positive modulator GS39783 reverse some behavioural changes related to positive syndromes of psychosis in mice.
Br J Pharmacol.
2011; 163(5): 1034–47. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 244.
Wierońska JM, Kłeczek N, Woźniak M, et al.:
mGlu5-GABAB interplay in animal models of positive, negative and cognitive symptoms of schizophrenia.
Neurochem Int.
2015; 88: 97–109. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 245.
Turrigiano GG:
The dialectic of Hebb and homeostasis.
Philos Trans R Soc Lond B Biol Sci.
2017; 372(1715): pii: 20160258. PubMed Abstract
| Publisher Full Text
| Free Full Text
Comments on this article Comments (0)