Keywords
python, bioinformatics, NGS, DNA, pedigree-information, gene-drop, gene-burden, kinship, mapreduce
This article is included in the Bioconductor gateway.
This article is included in the University College London collection.
This article is included in the Python collection.
python, bioinformatics, NGS, DNA, pedigree-information, gene-drop, gene-burden, kinship, mapreduce
Seqfam is a python package which is most useful in analysing next generation sequencing (NGS) DNA data from families with known pedigree information in order to identify rare variants that are potentially causal of a disease/trait. It was originally developed to analyse the whole exome sequencing data of a cohort of 200 families affected by a particular complex disease. Family-based study designs are often used to identify rare causal variants because such variants may be substantially more frequent in affected families than the general population (Auer & Lettre, 2015). Seqfam contains modules for Monte Carlo gene dropping (gene_drop.py) (MacCluer et al., 1986), flagging variants based on their pattern of occurrence within families (pof.py), verification of ascertained pedigrees via kinship coefficients (relatedness.py), regression-based gene burden testing (gene_burden.py), and an additional module that facilitates the creation of job submission scripts on a computer cluster (sge.py).
For a rare variant to be considered potentially causal of a particular trait/disease based on in silico analysis, it must satisfy various criteria, such as being biologically plausible and predicted to be pathogenic. The user can run analyses with the gene_drop.py and pof.py modules to acquire additional evidence. Given the structure of the families, Monte Carlo gene dropping can assess whether a variant is enriched in the cohort relative to the general population, and assuming the trait/disease is more prevalent in the cohort, such enrichment supports causality. The user can use the pof.py module to identify variants which are carried by most or all affected members of a family, or even which segregate between affected and unaffected members. The authors are unaware of existing software packages for performing these analyses in familial cohorts, so gene_drop.py and pof.py fulfil this need. The gene_drop.py module can be considered complementary to the RVsharing R package (Bureau et al., 2014), which calculates the probability of multiple affected relatives sharing a rare variant under the assumption of no disease association or linkage.
The gene_burden.py module implements the Combined Multivariate and Collapsing (CMC) burden test (Li & Leal, 2008) for detecting rare causal variants, where the multivariate test is a log-likelihood ratio test. The user can supply covariates to control for potential confounders such as divergent ancestry. This burden test should be applied to unrelated samples, and hence is of no use for cohorts containing few families. However, for cohorts containing a relatively large number of families, a sufficient number of unrelated cases can be extracted and combined with a separate set of unrelated controls. Burden tests aggregate rare variants in a gene or functional unit into a single score (Li & Leal, 2008; Madsen & Browning, 2009; Morris & Zeggini, 2010; Price et al., 2010), and are one broad class of statistical methods which combine the effects of rare variants in order to increase power over single marker approaches. Sequence kernel association testing (SKAT) (Wu et al., 2011) is another widely-used sub-category of such methods. In general, burden testing is more powerful than SKAT when a large proportion of variants are causal and are all deleterious/protective.
Stand-alone software exists for performing CMC testing with covariates e.g. RVTESTS (Zhan et al., 2016) and PLINK/SEQ, but gene_burden.py is an attractive alternative due to its use of the versatile Pandas library. The CMC test function takes malleable Pandas data frames as input/output, which gives the user great flexibility in pre/post-processing the data, and this in turn may reduce I/O overhead. For example, let us consider variant grouping (e.g. by gene) and aggregation. This is specified via columns in the data frame, so the user can experiment widely by modifying them in python code e.g. group variants by functional unit instead of gene, aggregate within alternative sets of population allele frequency ranges, include/exclude unaggregated variants, and even aggregate by another factor such as consequence. As well as this flexibility, the function also produces rich output, including information about the independent variables.
The potential for genetic discovery in DNA sequencing data is reduced when samples are mislabelled. Hence, necessary quality control steps include identifying duplicates, and in the case of familial samples, verifying the ascertained familial relationships described in the pedigrees. The relatedness.py module facilitates these quality control steps and is used in conjunction with KING software (Manichaikul et al., 2010). Given genotypes for relatively common variants, KING can efficiently calculate a kinship coefficient for each sample pair. The relatedness.py module can then map each kinship coefficient to a degree of relationship and check it corresponds with the pedigree. KING is often already part of NGS analysis pipelines, so incorporating relatedness.py is straightforward. Peddy (Pedersen & Quinlan, 2017) is an alternative which does not require KING.
The final module, sge.py, has general utility in running analyses of NGS data (and indeed any “big data”) on computer clusters. Many NGS data analyses can be cast as “embarrassingly parallel problems” and hence executed more efficiently on a computer cluster using a “MapReduce pattern”: the overall task is decomposed into independent sub-tasks (“map” tasks), then the map tasks run in parallel and after their completion, a “reduce” action merges/filters/summarises the results. For example, gene burden testing across the whole exome can be decomposed into independent sub-tasks by splitting the exome into sub-units e.g. chromosomes. Sun Grid Engine (SGE) is a widely used batch-queueing system, and analyses can be performed in a MapReduce pattern on SGE via so-called array jobs. The sge.py module can be used to automatically create the scripts required for submitting and running an array job.
This section describes the functionality and methods employed by seqfam’s 5 modules, which are:
1. gene_drop.py: Monte Carlo gene dropping;
2. pof.py: variant pattern of occurrence in families;
3. gene_burden.py: regression-based gene burden testing;
4. relatedness.py: identification of duplicates and verification of ascertained pedigree information via kinship coefficients;
5. sge.py: Sun Grid Engine (SGE) array job creation.
Figure 1 provides a visual representation of modules 1–4.
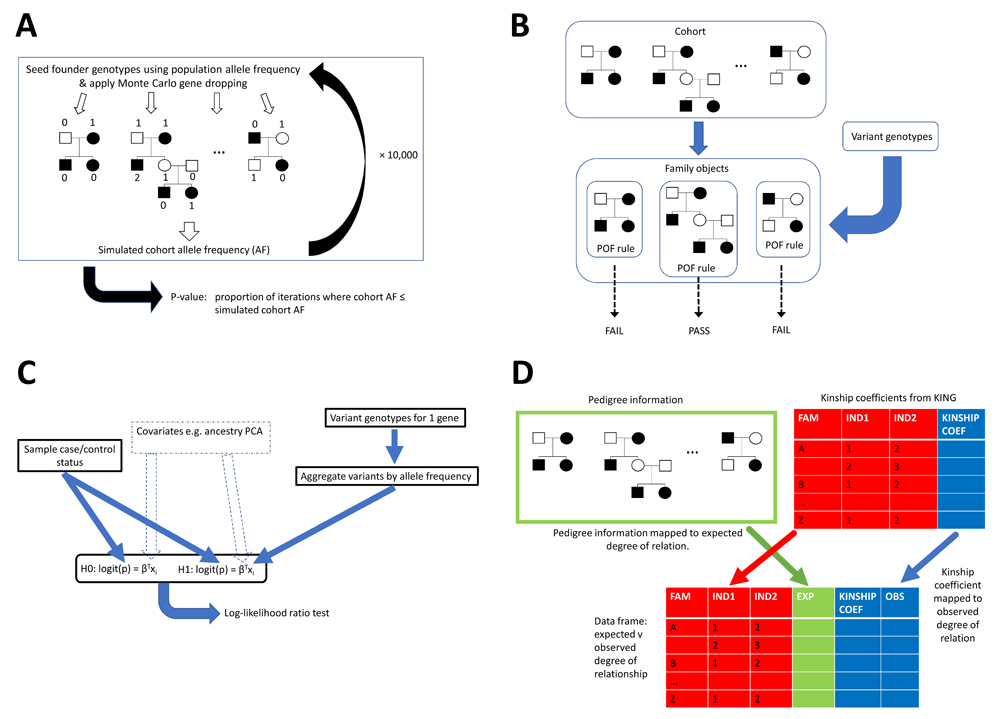
Panel A represents the Cohort.gene_drop method in the gene_drop.py module which performs Monte Carlo gene dropping. On a single iteration, for each family the algorithm seeds founder genotypes based on the variant population allele frequency and then gene drops via depth-first traversals. Having done this for all families, a simulated cohort allele frequency (AF) is calculated and following many iterations (e.g. 10,000), a p-value, the proportion of iterations where cohort AF < simulated cohort AF, is outputted. Panel B represents the Pof.get_family_pass_name_l method in the pof.py module. Prior to calling the method, each family is assigned a variant pattern of occurrence in family (POF) rule. The method then takes a variant’s genotypes and returns families whose POF rule is passed. Panel C represents the CMC.do_multivariate_tests method in the gene_burden.py module. This method takes sample affection status and variant genotypes across multiple genes, plus optionally covariates such as ancestry PCA coordinates. For each gene, the method aggregates the variants by allele frequency, constructs null and alternative hypothesis logit models which may include the covariates, and then performs a log-likelihood ratio test. Panel D represents the Relatedness.get_exp_obs_df method in the relatedness.py module. For input, this takes pedigree information and kinship coefficients from KING for each within-family sample pair. It maps these data to expected and observed degrees of relationship respectively, returning a data frame.
Gene dropping. By default, for each variant of interest, the gene_drop.py module performs 10,000 iterations of gene dropping in the familial cohort. In each iteration it gene drops in each family once, seeding the founder genotypes based on the population allele frequency. It then calculates the resulting simulated cohort allele frequency from samples specified by the user i.e. those which were sequenced. After completing all iterations of gene dropping, gene_drop.py outputs the proportion of iterations in which the true cohort allele frequency is less than or equal to the simulated cohort allele frequency: a low proportion, e.g. < 5%, is evidence of enrichment.
The module gene drops in a family in the following way. First, it assigns a genotype (number of copies of the mutant allele) to each founder using a Binomial distribution where the number of trials is 2 and the probability of success in each trial is the population allele frequency. Hence the founders are assumed to be unrelated. It then performs a depth-first traversal starting from each founder (1 per spousal pair), and for heterozygotes, uses a random number generator to determine which parental allele to pass onto the child. Thus, every individual in the family is assigned a genotype.
Variant pattern of occurrence in families. For each family, the user can use the pof.py module to define a variant pattern of occurrence rule and check whether any supplied variants pass. The rule can specify a minimum value for the proportion of affected members (As) who are carriers (A.carrier.p), and/or a minimum difference between the proportion of As and unaffected members (Ns) who are carriers (AN.carrier.diff). Constraints for the number of genotyped As and Ns can also be added.
As an illustrative example, consider a cohort in which families can be categorised as follows based on their number of As and Ns:
For the A4N1 families, the user may be interested in variants carried by all As and so require A.carrier.p = 1, while for the A3N2 families, they may be interested in variants which are more prevalent in As than Ns and so require AN.carrier.diff ≥ 0.5.
Gene burden. To use the gene_burden.py module, the user must first read the various required data into Pandas data frames. These data include variant annotations by which to group (e.g. gene/functional unit) and aggregate (e.g. population allele frequency), and the genotypes, affection status and covariates for the unrelated samples. The user can specify multiple categories in which to aggregate variants (e.g. into population allele frequency ranges of 0–1% and 1–5%), and variants outside these categories (e.g. more common variants) remain unaggregated. An aggregated variant category takes the value 0 or 1. For each variant group, having aggregated the variants, gene_burden.py will perform a multivariate test, which is a log-likelihood ratio test based on Wilk’s theorem:
Χ2=2(llh0 − llh1); df = dfh1 − dfh0
where ll is log-likelihood, h1 is the alternative hypothesis, h0 is the null hypothesis and df is degrees of freedom. Specifically, it is a log-likelihood ratio test on null and alternative hypothesis logit models where the dependent variable is derived from affection status, the variant variables (aggregated and/or unaggregated) are independent variables in the alternative model and the covariates are independent variables in both. The logit models are fitted using the Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm.
Since aggregation by population allele frequency is common, the module provides a function to assign variants to allele frequency ranges based on multiple columns (derived from different population databases such as gnomAD (Lek et al., 2016)). The user specifies a preference order in case the variant is absent from the most preferred database(s).
Relatedness. As input, the relatedness.py module requires pedigree information and a file containing kinship coefficients outputted by KING. For each sample pair, the module will map the pedigree information and kinship coefficient to an expected and observed degree of relationship respectively. The mapping from kinship coefficient to relationship is as specified in KING documentation: > 0.354 for duplicate samples/monozygotic twins, 0.177–0.354 for 1st degree relatives, 0.0884–0.177 for 2nd degree relatives, 0.0442–0.0884 for 3rd degree relatives, and < 0.0442 for unrelated. The user can change this mapping if they wish.
Computer cluster array job creation. To use the sge.py module, the user must first create lists of map tasks, map tasks requiring execution and reduce tasks. The map tasks requiring execution are map tasks which have not previously completed successfully and hence need to run. Given these lists, the sge.py module can create all necessary scripts/files for submitting and running an array job. This includes scripts for all map tasks, a text file specifying that only the map tasks requiring execution should run, and a master executable submit script for submitting the array job to the job scheduler.
Seqfam is compatible with Windows, Mac OS X and Linux operating systems. It is coded using Python 3.6, but can also be run by Python 2.7. It requires various data analysis libraries/packages, almost all of which can be acquired by downloading and installing the Anaconda python distribution. The StatsModels module, which is not in the Anaconda distribution, is also required. Having cloned the repository, the user should add the repository src directory to their environmental python path.
The repository contains additional scripts in src/examples which demonstrate the functionality of the modules on example data, including files in the data directory. The scripts are 1_test_gene_drop.py, 2_test_pof.py, 3_test_gene_burden.py, 4_test_relatedness.py, and 5_test_sge.py. The reader can also refer to Table 1 for a summary of the main user functions of the 5 seqfam modules, which includes their input/output. Data in the example data files are derived from the whole exome sequencing of a large cohort of over 200 families with inflammatory bowel disease (unpublished study1).
tsv = tab-separated values; AF = allele frequency; POF = pattern of occurrence in family.
The only input file for 1_test_gene_drop.py is cohort.tsv, which contains the pedigree information for an example familial cohort. This cohort has 3,608 samples from 251 families, and the complexity of the families, calculated as 2n-f where n and f are the number of non-founders and founders respectively (Abecasis et al., 2002), has median 9 and range 0–103. The cohort.csv file is in fam file format (Purcell et al., 2007), meaning it has 1 row per individual and 6 columns for family ID, person ID, father, mother, sex and affection.
The 1_test_gene_drop.py script first creates a Cohort object from cohort.tsv, which stores each family tree, then calls the gene_drop method with the following arguments: pop_af and cohort_af are the allele frequency of a particular variant in the general population and cohort respectively, sample_genotyped_l is the list of cohort samples with a genotype, and gene_drop_n is the number of iterations of gene dropping to perform. Hence the samples in sample_genotyped_l are used by the user to calculate cohort_af, and by the method to calculate the simulated cohort allele frequencies. The method returns a p-value. The script calls the gene_drop method with ascending values for cohort_af, and so descending p-values are returned.
In benchmarking with the Unix time command on an Intel Xeon CPU E7-4830 v2 processor (20M Cache, 2.20 GHz), creating a cohort object and then executing a single call of the gene_drop method (gene_drop_n=1,000) required approximately 25 seconds of Computer Processing Unit (CPU) time. CPU time scales linearly with gene_drop_n and cohort size.
There are no input files for 2_test_pof.py. The example script first creates a Pof object which stores a couple of Family objects, each representing an example family and its variant pattern of occurrence rule. Next it calls the Pof object’s get_family_pass_name_l method with the argument genotypes_s, which is a Pandas Series containing sample genotypes for a particular variant. The method returns a list of families whose pattern of occurrence rule is passed by this variant.
The 3_test_gene_burden.py script uses the gene_burden.py module to perform CMC tests on example data: it performs 1 CMC test per gene where variants in the population allele frequency ranges of 0-1% and 1-5% are aggregated, and any variants ≥ 5% remain unaggregated. The input files are in the data/gene_burden directory: samples.csv, genotypes.csv and optionally covariates.csv. The samples.csv file contains the samples’ ID and affection status where 2 indicates a case and 1 a control, and genotypes.csv contains 1 row per variant with columns for the sample genotypes, and for variant grouping and aggregation e.g. gene and population allele frequency. A sample’s genotype is the number of alternate alleles which it carries (0-2). The covariates.csv file contains the covariates to control for, which in this case are ancestry PCA coordinates.
The script first reads samples.csv into a Pandas Series, and genotypes.csv and covariates.csv into Pandas DataFrames. These data frames are indexed by variant ID and covariate name respectively. Having created a CMC object, the script calls its assign_vars_to_pop_frq_cats method in order to map the variants to the desired population allele frequency ranges. Multiple population allele frequency columns (databases) are used here, ordered by descending preference. The mapping is stored in a new column in the genotypes data frame. Finally, the script calls the do_multivariate_tests method to perform the CMC tests, specifying the gene column for grouping the variants, and the new allele frequency range column for aggregation. The results are written to a CSV (comma-separated values) file and returned in a data frame. They include the number of variants in each aggregation category, the number of unaggregated variants (“unagg” column), the log-likelihood ratio test p-value with/without covariates (“llr_p” and “llr_cov_p”), and the coefficient/p-value for each aggregated variant variable (“_c” and “_p”).
The input files for 4_test_relatedness.py are cohort.tsv (as used in gene dropping), and data/relatedness/king.kinship.ibs which was outputted by KING and contains kinship coefficients for within-family sample pairs. The example script first creates a Relatedness object which stores the paths to these files, then calls the object’s find_duplicates and get_exp_obs_df methods. The former returns any within-family sample duplicates, and the latter returns a Pandas DataFrame containing the expected and observed degree of relationship for each within-family sample pair. Finally, the script prints the sample pairs which have a different expected and observed degree of relationship.
There are no input files for 5_test_sge.py. This script first makes lists of map tasks (map_task_l), map tasks to execute (map_task_exec_l), and reduce tasks (reduce_task_l). Here map_tasks_exec_l contains every other map task. Next, the script creates an SGE object which stores the directory where job scripts will be written (here data/sge). Finally, it calls the object’s make_map_reduce_jobs method with the following arguments: a prefix for all job script names (here “test”) and the above 3 lists. This writes the job scripts, and were they for a real array job (they are not), the user could then submit it to the job scheduler by running the master executable submit script data/sge/submit_map_reduce.sh. The test.map_task_exec.txt file specifies which map tasks to run i.e. the map tasks in the map_tasks_exec_l list.
This article has introduced seqfam, a python package, primarily designed for analysing NGS DNA data from families with known pedigree information in order to identify rare variants that are potentially causal of a disease/trait of interest. It currently includes modules for verification of pedigree information, gene dropping, applying variant pattern of occurrence rules in families, gene burden testing and job script generation on a computer cluster.
Latest source code and example files are available at: https://github.com/mframpton/seqfam
Archived source code as at time of publication: http://doi.org/10.5281/zenodo.1173768 (Frampton, 2018)
License: GNU General Public License v3.0
1Authors involved in the unpublished study: E. R. Schiffa, M. Framptona, F. Semplicia, N. Pontikosb, S. Bloomc, S. McCartneyc, R. Vegac, L. Lovatd, E. Woode, A. Hartf, D. Crespig, M. Furmang, S. Mannh, C. Murrayi, A. P. Levinea and A. W. Segala
aCentre for Molecular Medicine, Division of Medicine, University College London
bInstitute of Ophthalmology, Moorfields Eye Hospital, University College London
cDepartment of Gastroenterology. University College London Hospital
dResearch Department of Tissue and Energy, Division of Surgery and Interventional Science, UCL, U.K.
eGastroenterology Department, Homerton University Hospital, London, U.K.
fGastroenterology Department, St Marks Hospital, U.K.
gCentre for Paediatric Gastroenterology, Royal Free Hospital, London, U.K.
hGastroenterology Department, Barnet General Hospital, U.K.
iCentre for Gastroenterology, Royal Free Hospital, London, U.K.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)