Keywords
tick, genome, cell line, ISE6, Ixodes scapularis
This article is included in the Genomics and Genetics gateway.
tick, genome, cell line, ISE6, Ixodes scapularis
The Ixodes scapularis embryonic 6 (ISE6) cell line is a widely used resource that is permissive to pathogens including human pathogens transmitted by ticks. Two decades ago, a collection of I. scapularis cell lines were derived from embryonated eggs including IDE lines derived from northern ticks and ISE lines derived from southern ticks (Munderloh et al., 1994). Recent proteomics analysis suggested that the ISE6 line is derived from neuronal cells (Oliver et al., 2015). ISE6 cells have been used to isolate and analyze bacterial pathogens including: the causative agent of human granulocytic ehrlichiosis (HGE) (Munderloh et al., 1999); Borrelia burgdorferi, the causative agent of Lyme disease (Obonyo et al., 1999); the causative agent of southern tick-associated rash illness (STARI) (Varela et al., 2004); and Rickettsia felis, the causative agent of spotted fever (Pornwiroon et al., 2006). ISE6 cells have been used to study viral pathogens including: Semliki Forest virus (SFV) and Hazara virus (arbovirus, family Bunyaviridae, genus Nairovirus) (Garcia et al., 2005); and Langat virus (LGTV), a Flavivirus (Grabowski et al., 2016). The cells have also been used to study RNAi and genome engineering in ticks; reviewed in (Oliver et al., 2015).
The Ixodes scapularis (black-legged tick) genome had been estimated to harbor 70% repeat content (Ullmann et al., 2005) when our lab participated in a community effort to sequence and assemble a tick genome. A reference sequence built from 3.8X Sanger sequencing and the Celera Assembler (Istrail et al., 2004; Myers et al., 2000) software was fractured into 570,640 contigs (369,495 scaffolds) with a contig N50 of only 2,942 bp. The total contig span was 1.388 Gbp though the genome size was estimated at 2.1 Gbp. The assembly supported the annotation of 20,486 protein-coding genes and an extensive analysis of tick biology (Gulia-Nuss et al., 2016). The assembly is maintained at VectorBase (Giraldo-Calderón et al., 2015) under the name IscaW1.
A genome assembly for the ISE6 genome would assist investigations of ISE6 as a biological system. It would also provide a host subtraction tool for ISE6-based sequencing assays. Host subtraction is the bioinformatics process of filtering reads whose origin is host DNA and RNA (Daly et al., 2015). Host subtraction enriches the non-host component of sequence datasets and is especially attractive for assays involving high-throughput sequencing technologies that generate short reads in high volume where data reduction can realize cost savings. Following host subtraction, remaining reads can be mapped to references and counted, or used as queries to sequence databases, or assembled to reconstruct novel transcript or genome sequences. With an expectation that the IES6 genome would be as challenging as the tick genome, we sequenced IES6 with high coverage and long reads that, taken together, might generate a high quality reference genome assembly.
ISE6 cells were obtained from the American Type Culture Collection (ATCC), cell line CRL-11974, lot number 100005, patent 5,869,335. This cell line had been isolated from ticks collected in Georgia, USA. As generally described by ATCC technical bulletins and personal communications, cells were grown in Leibovitz L-15B media pH 7.0 (Leibovitz, 1963) (ThermoFisher Scientific) supplemented with 80 mM glucose, 10% tryptose phosphate broth, 0.1% bovine lipoprotein cholesterol concentrate and 2% heat inactivated fetal bovine serum (Munderloh & Kurtti, 1989). An addition of 0.7% non-essential amino acids (NEAA) concentrate (ThermoFisher Scientific) was also added prior to incubation. The cells were incubated in a gently shaking flask at 31°C with no C02.
For short-read sequencing, genomic DNA was isolated from the cell line using a Qiagen genomic DNA isolation kit. Bioanalyzer analysis confirmed high molecular weight DNA was recovered. The library was size selected using Pippin Prep and prepared using the NextGen paired end barcoded genomic library construction protocol. Library quantification and normalization was performed by qPCR. The library was sequenced on the Illumina NextSeq 500 platform to generate 2x150 paired reads. Reads were demultiplexed which removed barcodes and sequencing adapters, and further treated with CutAdapt 1.8.1 to remove any remaining adapter.
For long-read sequencing, cells were grown until they attached to the flask. Genomic DNA was extracted from ISE6 cells using a Qiagen Genomic DNA isolation kit, stopping prior to the G2 isolation step. Frozen pellets and frozen cells were shipped to the Icahn School of Medicine at Mount Sinai for library construction and sequencing using standard SMRTbell template preparation kits (Pacific Biosciences). A total of 52 SMRTcells were run on the PacBio Sequel platform using standard PacBio protocols.
The long reads were corrected and assembled with the Canu assembler (Koren et al., 2017) version 1.6. Canu was run with the SGE grid engine and Java 1.8 using default parameters except: minOverlapLength = 1000 bp, corMhapSensitivity = "low", and genomeSize = 1 Gbp. The contig consensus sequences were polished using SMRT Link version 5.0.1.9585 which includes Arrow version 2.2.1, blasr 5.3, and pbalign 0.3.1 (Pacific Biosciences).
Short reads were mapped with bowtie2 (Langmead & Salzberg, 2012) version 2.2.5 using either end-to-end or local-alignment mode as indicated in the text. Using default settings, the mapper reported at most one mapping per read and reported read maps individually though it used reads as pairs to select alignments. Mappings were analyzed with samtools (Li et al., 2009) version 1.2.1 and bedtools (Quinlan, 2014; Quinlan & Hall, 2010) version 2.26. K-mers were counted using Jellyfish (Marçais & Kingsford, 2011) version 2.2.6 for several values of K: 11, 15, 21, 41, 51, and 61. Each computed K-mer histogram was analyzed with GenomeScope (Vurture et al., 2017). Single-copy gene analysis used BUSCO (Simão et al., 2015) version 3.0.2 with Arthropoda OrthoDB version 9. Alignments between the tick and cell line assemblies were computed on contigs from each assembly using nucmer, the local aligner in the MUMmer package (Kurtz et al., 2004) version 3.1. The BLAST analysis used TBLASTN in NCBI BLAST+ (Camacho et al., 2009) version 2.2.31. The Ixodes protein predictions were downloaded from UniProt in Nov 2017.
PacBio sequencing yielded 192.5 Gbp in 27.3 million unpaired reads, providing approximately 92X coverage of the estimated 2.1 Gbp tick genome. This dataset included 190.7 Gbp in reads >= 1 Kbp and 115.9 Gbp in reads >= 10 Kbp. To overcome high base call error observed in single-molecule long-read data, the long reads were subjected to the Canu correction process which filtered, trimmed, and polished reads based on alignment with other long reads (Koren et al., 2017). Correction yielded 36.7 Gbp in 2.1 million reads with a 17,680 bp N50. Read counts are shown in Table S1. Read length distributions, before and after correction, are shown in Figure S1.
The corrected long reads were assembled in isolation with the Canu long-read assembler (Koren et al., 2017). The initial assembly, named Ise6_asm0, contained 18,717 contigs. The uncorrected long reads were used to polish the contig consensus sequences using the Arrow process. This was run in two iterations to produce assemblies Ise6_asm1 and Ise6_asm2 respectively. The released assembly, named Ise6_asm2, contained 2,691,078,110 bases in 18,717 contigs with a 269,660 bp contig N50. Statistics for each assembly are shown in Table S2.
Since the tick contigs are generally smaller than the cell line contigs, it seemed likely that some tick contigs would be wholly contained by cell line contigs. The IscaW1 and Ise6_asm0 assemblies were compared with the MUMmer local alignment software; see Table S3. A high-confidence alignment subset (filtered with delta-filter -1) was used. The average identity of aligned bases was 96.58%. Both assemblies were almost fully covered by alignments. Using alignments of 1 Kbp or more, IscaW1 contigs with at least one alignment made up 78.6% of IscaW1 bases. The set of IscaW1 contigs with at least 90% coverage in any one alignment made up 35.5% of IscaW1 bases.
To provide an orthogonal dataset for assembly assessment, the genome was also sequenced on a short-read platform. Illumina sequencing yielded 25 Gbp in 171 million 2x150 bp paired reads, providing approximately 24X short-read coverage of the 2.1 Gbp estimated genome size of the tick. Comparative map statistics are shown in Table S4. When paired reads were mapped to Ise6_asm0 using a local alignment algorithm, 97.83% of reads mapped as a concordant pair. For comparison, when the reads were mapped to the IscaW1 tick reference assembly, 91.68% of reads mapped as a concordant pair. This result indicates that the ISE6 assembly is more representative of ISE6 genome structure than the Ixodes reference.
For consensus quality assessment, the paired reads were mapped to Ise6_asm2 and IscaW1 contigs using a more stringent, global (end-to-end) alignment algorithm; see Table S4. Among these alignments, the read sequence disagreement with the contig consensus was 1.79% for Ise6_asm2 and 5.03% for IscaW1. This demonstrates that the ISE6 consensus is more representative of ISE6 genome sequence than the Ixodes reference.
The rates of concordant pair mapping to zero, one, or multiple sites were 23%, 29%, and 48% respectively for Ise6_asm2 and 44%, 30%, and 25% for IscaW1; see Table S4. Thus, by paired-read mappability, both assemblies contain 29%–30% unique sequence while the ISE6 assembly captures an additional 23% of reads and these align to repeat sequences in the assembly.
The global alignment 23% unmapped rate in Ise6_asm2 is an order of magnitude larger than the unmapped rate among the local alignments. It is possible that the long and short read sequencing captured genuine differences at unstable regions of the cell line genome. It seems more likely that the genome harbors repeat instances that are similar-but-not-identical to those in the assembly.
Using the global alignments and accepting all mapped reads (whether mapped as a pair or not), the Ise6_asm2 assembly mapped 81% of reads while IscaW1 mapped 65%. Thus, the Ise6_asm2 assembly outperformed the IscaW1 assembly as a host subtraction tool using pairwise local, pairwise global, and read-wise global alignments.
The assembly was assessed for completeness using gene content analysis. The latest UniProt protein predictions on the IscaW1 tick genome assembly were used as TBLASTN query sequences against the cell line assembly. Out of 20,473 predicted proteins: 20,290 (99.1%) had at least one hit in Ise6_asm0 while 183 predictions had no hit. The Ise6_asm2 assembly was analyzed for gene content using the BUSCO collection of genes thought to be single-copy in arthropod genomes; Table S5. Of 1066 genes searched, 1.4% were fragmented, 3.6% were missing, and 95% were complete. These results indicate that the assembly is fairly complete for single-copy genes.
The Ise6_asm2 contig span is 2.8 Gbp which exceeds the 1.4 Gbp contig span of the IscaW1 tick reference assembly as well as the 2.1 Gbp estimated genome size for tick. The discrepancy could be due to several factors. It is possible that the cell line genome is larger than the tick genome, or that the assembly contains dual representations of heterozygous loci that assembled separately, or that the IscaW1 reference assembly underrepresents repeats present in the tick and ISE6 genomes. These possibilities were explored with several analyses.
K-mer analysis (Vurture et al., 2017) provides an assembly-free genome size estimate extrapolated from the frequency distributions of short, contiguous sequences extracted from the sequencing reads and counted using an exact-match algorithm (Marçais & Kingsford, 2011). In K-mer analysis of our short-read data, the observed distribution could not be fit to a model. Analysis of the corrected long read K-mers was similarly inconclusive. As illustrated by the representative plot in Figure S2, the distribution does not include a strong peak other than the mode at 1X. These results are possibly due to the low coverage in both datasets, which would be especially low if heterozygous haplotypes were represented separately in contigs.
Next, genome size estimation was attempted using coverage analysis of paired reads mapped to contigs. Pairs mapped end-to-end provided 39.9 Gbp of mapped bases. In the distribution of read coverage per contig base, there is a smooth peak with a 9X mode and a tail at higher coverage; see Figure S3. Using the mapped base count divided by 9X to represent the average coverage of unique sequence in the genome, the extrapolated genome size is 4.43 Gbp, which is about twice as large as the tick estimated genome size. This suggests that the assembly process separated the haplotypes of a 2.22 Gbp diploid genome.
Long-read coverage was analyzed next. The uncorrected long reads had been mapped to Ise6_asm1 contigs for the final iteration of Arrow consensus polish. Based on those results, the per-base coverage peaks at 34X with a shoulder at almost twice that level; see Figure 1. This suggests that 34X represents the coverage mode for haplotype-separated sequence. With 175.48 Gbp in mapped reads, 158.47 Gbp of read sequence aligned to 151.93 Gbp of contig sequence. These mapping and coverage results combine to indicate 151.93 / 34 = 4.47 Gbp size for the combined haplotypes (diploid) and 2.24 Gbp for the haploid genome. Thus, the completeness of the 2.8 Gbp assembly is uncertain and the assembly appears to harbor double-representation of loci that are haplotype-separated and under-representation of genomic repeats, as indicated in Figure 1.
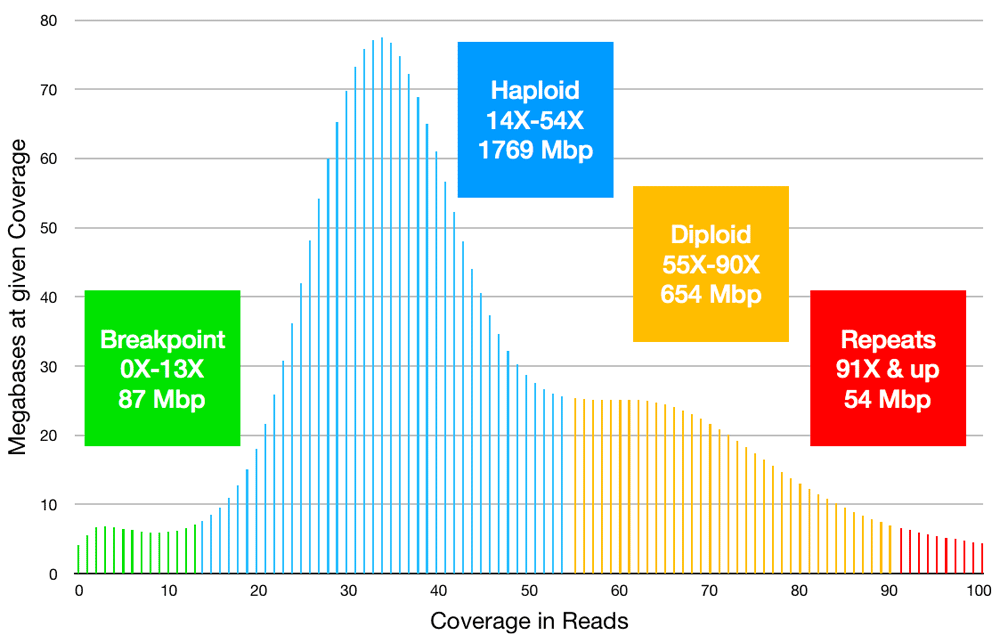
Read coverage per base was computed by mapping uncorrected long reads to assembled contigs. Colors were added to highlight the following interpretation. Green: the minor peak of low-coverage bases suggests 87 Mbp of contig bases lies near breakpoints such as contig ends and false joins. Blue: the dominant peak, with mode at 34X, suggests that 1769 Mbp of the assembly is haplotype-separated sequence possibly representing 885 Mbp of the diploid genome. Yellow: the shoulder near 64X suggests that about 654 Mbp of the diploid genome is captured as diploid-consensus sequence. Red: high coverage (including bases not shown with coverage over 100X) suggests that under-represented genomic repeats occupy 54 Mbp of the assembly but more of the genome.
Local alignments between assemblies were examined for support of the haplotype separation hypothesis. The cell line Ise6_asm0 contigs were aligned to the tick reference IscaW1 contigs using the nucmer local alignment software. As shown in Figure S4, the largest IscaW1 contig aligns full-length to two contigs of Ise6_asm0. The full set of alignments was filtered to retain one best alignment per Ise6_asm0 position (with delta-filter -q) and to retain only IscaW1 contigs with at least 50% coverage in one such alignment. Using these alignments to distinguish IscaW1:ISE6 multiplicities, 43% of IscaW1 bases are in contigs with 1:1 relation to ISE6, while 17% of bases are in contigs with 1:2 relations, and 3% of bases are in contigs with relations of 1:3 or higher; see Table S3. Not all contigs could be categorized. Further work would be required to fully partition and phase all duplicated contigs with high confidence.
Of 1066 BUSCO genes searched against Ise6_asm2, 67% were complete and single-copy, and 28% were complete and duplicated. This indicates that almost a third of single-copy genes are duplicated in the assembly, presumably due to haplotype separation. If this rate of sequence duplication applies to the assembly overall, the 2.67 Gbp assembly indicates a 2.29 Gbp genome size.
During the assembly submission to GenBank, NCBI flagged one contig for sequence similarity to Rickettsia, a genus of endosymbionts common to Ixodes scapularis (Gillespie et al., 2012; Labruna et al., 2007; Steiner et al., 2008; Zeringóta et al., 2017). The long read coverage in the flagged region differs from the flanking regions; see Figure S5. The contig was split and re-submitted without the flagged region.
The ISE6 cell line, derived from the Lyme disease tick Ixodes scapularis, is widely used but has so far lacked a genome reference sequence. With this report, the genome has been sequenced, assembled, and released. The genome sequence was generated from high coverage in PacBio Sequel long reads, assembled with the Canu assembler, and polished with Arrow. Gene content analysis indicated that the assembly is largely complete though read mapping analysis indicated that some genomic repeats are under-represented in the assembly. Read mapping and single-gene analysis indicated that portions of the genome are represented as a diploid consensus while other portions are represented in haplotype-separated copies. The new assembly provides a more accurate representation of the cell line genome compared to the previous closest reference, which was an assembly of the I. scapularis tick genome. In our mapping of cell line gDNA short read pairs that were not used for the assembly, the cell line assembly was more effective for identifying host reads compared to the tick reference. Thus, the new assembly provides a resource for analysis of the cell line and for host subtraction to assist the detection of pathogens present in the cells.
Comparable genome size estimates were obtained by three methods. Short-read coverage analysis indicated 2.22 Gbp. Long-read coverage indicated 2.24 Gbp. Single-copy gene analysis indicated 2.29 Gbp. The tick genome was previously estimated to be 2.1 Gbp so the cell line may harbor some ISE6-specific sequence. Identification of such sequences is left for future work. Our local alignments of the tick and ISE6 assemblies covered nearly all of both assemblies, so any cell-line-specific sequence is likely to involve genomic repeats.
We hope to enhance the assembly resource in several ways. We hope to generate a second version of the sequence using different assembly parameters. A larger genome size estimate, in particular, should lead to higher generated coverage in corrected reads. Additional corrected reads would necessarily be shorter than the ones used here, but they could provide additional depth for repeat detection and repeat resolution. We hope to provide gene annotation, repeat content analysis, and more specific haplotype separation analysis of the second assembly.
The gDNA Illumina and PacBio reads are available at NCBI SRA under BioSample SAMN06329993. This entire assembly has been deposited at GenBank under the accession GCA_002892825.1. Whole Genome Shotgun sequencing project is available under accession PKSA00000000.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)