Introduction
Historically, mtDNA sequence has been used in taxonomy as a source of species diagnostic markers (Cronin et al. (1991); De Barba et al. (2014); Pegg et al. (2006)) or in population genetics and evolutionary studies (Fu et al. (2013); Harrison (1989); Llamas et al. (2016)). One advantage of using mitochondrial over nuclear DNA for such studies is that the mutation rate of mtDNA is about 10 times faster than nuclear DNA (Brown et al. (1979); Haag-Liautard et al. (2008)), hence amplifying the evolutionary trajectory of populations and species. In addition, mtDNA is easy to amplify, because there are more copies of mitochondrial DNA relative to nuclear DNA. Also, universal primers can be applied to a wide range of species. Widely used universal primers target the cytochrome b and cytochrome oxidase 1 genes (Tahir et al. (2016)), because both have conserved and highly variable regions. In addition to these, other genes as described in De Mandal et al. (2014), can also be used as markers. However, phylogenetic trees based on mtDNA can deviate from the ones that are derived from nuclear DNA (Phillips et al. (2013); Shaw (2002); Sota & Vogler, 2001).
The Anopheles gambiae species complex consists of eight morphologically identical species that can only be distinguished with molecular markers (Scott et al. (1993); Coetzee et al., 2013) or, for some of the species, by cytological examination of polytene chromosomes (Green, 1972; Pombi et al., 2008). The currently used molecular markers to distinguish between An. coluzzii and An. gambiae (Lee et al., 2014) are located within genomic islands of divergence located proximal to the centromeres (Turner et al. (2005)). Monitoring additional species-specific markers on mitochondrial DNA (mtDNA) could increase the ease of application and accuracy of species detection assays. In addition, mtDNA markers could enhance our understanding of divergence times among taxa within the complex.
Previous studies showed that there is a high amount of interspecific gene flow in mtDNA between An. coluzzii, An. gambiae and An. arabiensis specimens (Besansky et al., 2003; Besansky et al., 1997; Donnelly et al., 2004). Although these data suggested no evidence for clear species division among the various species, the studies only focused on the ND5 loci (Besansky et al., 2003; Donnelly et al., 2004) or included also cytochrome b and ND1 loci (Besansky et al., 1997). In our study we use the complete mitogenome for comparison, which would make the analysis more robust. In addition, we specifically included the different chromosomal forms in our analysis. These chromosomal forms are genetically diverged from each other and display strong assortative mating in the An. gambiae chromosomal forms (Touré et al., 1998). The An. coluzzii chromosomal forms differ from each other in their ecology: An. coluzzii-Mopti is found in dry areas whereas the An. coluzzii-Forest restrtict themselves to a wet climate (Lee et al., 2009).
In this study we wished to identify species-specific markers within the mtDNA for Anopheles arabiensis, An. coluzzii and An. gambiae, including among the chromosomal forms currently subsumed under the designations An. gambiae and An. coluzzii, with the goal of adding these to our existing Anopheles species detection assay (Lee et al. (2014)). We sequenced the whole mitogenomes of 70 individual mosquito specimens collected throughout Sub-Saharan Africa. The raw Illumina sequencing reads were mapped to the AgamP4 reference sequence, which included both nuclear and mitochondrial sequences. We explore the relationship among An. arabiensis, An. coluzzii, An. gambiae and four of the sub-specific chromosomal form mitogenome sequences.
Methods
Sample collection
Anopheles arabiensis raw Illumina sequencing reads were obtained from our previous study (Marsden et al. (2014)). These included 20 female An. arabiensis mosquitoes which were collected indoors in houses using mouth aspirators from three villages in Tanzania in 2012 (Lupiro ((-8.38000°N, 36.66912°W), Sagamaganga (-8.06781°N, 36.80207°W), and Minepa (-8.25700°N, 36.68163°W) in the Kilombero Valley) and 4 samples from Cameroon collected in 2005 (9.09957°N, 13.72292°W). The DNA was extracted from the head and thorax of each mosquito species and An. arabiensis mosquitoes were identified using Scott primers (Scott et al., 1993)). The adult An. gambiae and An. coluzzii samples were collected indoors using mouth aspirators in Kela, Mali (11.88683°N, -8.44744°W) in 2012 and Mutengene, Cameroon (4.0994°N, 9.3081°W) in 2011. We subdivided the An. coluzzii specimen into the Forest and Mopti chromosomal forms. Similarly, we did this for the An. gambiae Savannah and Bamako chromosomal forms. We examined the polytene chromosome to characterize the chromosomal forms as in Lanzaro & Lee, 2013 and used the same definitions. The results of chromosome determination are listed in Table 1. The An. quadriannulatus mosquito, used as an outgroup for the phylogenetic analysis, was collected as larvae in the Shingwidzi area (23.1160°S 31.3752°E) in South Africa in 2015 and was reared to adult.
Table 1. List of detected chromosomal inversions to detect chromosomal forms of An. coluzzii and An. gambiae according Toure and co-workers (Touré et al., 1998). ‘2’ represents homozygous for the inversion, ‘1’ heterozygous for the inversion and ‘-‘ for homozygous for the standard arrangement.
| Banked ID | Chromosomal Form | 2La | 2Rb | 2Rc | 2Rd | 2Rj | 2Ru |
|---|
| 11MUTE470 | An. coluzzii-Forest | - | - | - | - | - | - |
| 11MUTE472 | An. coluzzii-Forest | - | - | - | - | - | - |
| 11MUTE476 | An. coluzzii-Forest | - | - | - | - | - | - |
| 11MUTE477 | An. coluzzii-Forest | - | - | - | - | - | - |
| 11MUTE479 | An. coluzzii-Forest | - | - | - | - | - | - |
| 11MUTE480 | An. coluzzii-Forest | - | - | - | - | - | - |
| 11MUTE483 | An. coluzzii-Forest | - | - | - | - | - | - |
| 11MUTE487 | An. coluzzii-Forest | - | - | - | - | - | - |
| 11MUTE490 | An. coluzzii-Forest | - | - | - | - | - | - |
| 11MUTE491 | An. coluzzii-Forest | - | - | - | - | - | - |
| 11MUTE493 | An. coluzzii-Forest | - | - | - | - | - | - |
| 2012KELA022 | An. coluzzii-Mopti | 1 | 1 | 1 | - | - | - |
| 2012KELA024 | An. coluzzii-Mopti | 2 | 1 | 1 | - | - | - |
| 2012KELA046 | An. coluzzii-Mopti | 2 | 1 | 1 | - | - | - |
| 2012KELA085 | An. coluzzii-Mopti | 2 | 2 | 2 | - | - | - |
| 2012KELA087 | An. coluzzii-Mopti | 1 | 2 | 2 | - | - | - |
| 2012KELA088 | An. coluzzii-Mopti | 2 | - | - | - | - | 1 |
| 2012KELA099 | An. coluzzii-Mopti | 2 | - | - | - | - | 1 |
| 2012KELA112 | An. coluzzii-Mopti | 2 | 2 | 2 | - | - | - |
| 2012KELA161 | An. coluzzii-Mopti | 2 | - | - | - | - | 1 |
| 2012KELA210 | An. gambiae-Savannah | 2 | 2 | - | - | - | - |
| 2012KELA214 | An. gambiae-Bamako | 2 | - | 2 | - | 2 | 2 |
| 2012KELA219 | An. gambiae-Bamako | 2 | - | 2 | - | 2 | 2 |
| 2012KELA228 | An. gambiae-Savannah | 2 | 2 | - | - | - | - |
| 2012KELA233 | An. gambiae-Savannah | 2 | 2 | - | - | - | - |
| 2012KELA234 | An. gambiae-Savannah | 1 | 2 | - | - | - | - |
| 2012KELA239 | An. gambiae-Bamako | 2 | 1 | 2 | - | 2 | 2 |
| 2012KELA240 | An. gambiae-Bamako | 2 | 1 | 2 | - | 2 | 2 |
| 2012KELA244 | An. gambiae-Bamako | 2 | - | 2 | - | 2 | 2 |
| 2012KELA285 | An. gambiae-Savannah | 2 | 2 | - | - | - | - |
| 2012KELA321 | An. gambiae-Savannah | 2 | 2 | - | - | - | - |
| 2012KELA334 | An. gambiae-Savannah | 2 | 2 | - | - | - | - |
| 2012KELA348 | An. gambiae-Savannah | 2 | 2 | - | - | - | - |
| 2012KELA367 | An. gambiae-Bamako | 2 | 1 | 2 | - | 2 | 2 |
| 2012KELA400 | An. coluzzii-Mopti | 2 | - | - | - | - | 2 |
| 2012KELA406 | An. gambiae-Bamako | 2 | - | 2 | - | 2 | 2 |
| 2012KELA409 | An. gambiae-Savannah | 2 | 2 | - | - | - | - |
| 2012KELA420 | An. coluzzii-Mopti | 2 | - | - | - | - | 2 |
| 2012KELA423 | An. coluzzii-Mopti | 2 | 2 | 2 | - | - | - |
| 2012KELA443 | An. gambiae-Bamako | 2 | 1 | 2 | - | 2 | 2 |
| 2012KELA457 | An. gambiae-Bamako | 2 | - | 2 | - | 2 | 2 |
| 2012KELA458 | An. coluzzii-Mopti | 2 | - | - | - | - | 2 |
| 2012KELA467 | An. gambiae-Bamako | 2 | - | 2 | - | 2 | 2 |
| 2012KELA468 | An. gambiae-Savannah | 2 | 1 | - | - | - | - |
| 2012KELA481 | An. gambiae-Bamako | 2 | 2 | 2 | - | 2 | 2 |
| 2012KELA496 | An. coluzzii-Mopti | 2 | 1 | - | - | - | - |
| 2012KELA651 | An. gambiae-Bamako | 2 | 2 | 2 | - | 2 | 2 |
| 2012KELA812 | An. gambiae-Savannah | 2 | 1 | - | - | - | - |
Genome sequencing
Sequencing methods for An. arabiensis samples are as described in Marsden et al. (2014). In short, individually barcoded Illumina paired-end sequencing libraries, with insert sizes of 320-400 basepairs (bp) using NEXTflex Sequencing kits (NOVA-5144) and barcodes (NOVA-514102)(Bio Scientific, Austin, TX, USA), were sequenced on an Illumina HiSeq2000 (Illumina, San Diego, CA, USA) with 100-bp paired-end reads using twelve samples per lane. For the An. coluzzii and An. gambiae samples we used the same methods as described in Norris et al. (2015) and Main et al. (2015). For the latter species, libraries were created using the Nextera DNA Sample Preparation Kit (FC-121-1031) and TruSeq dual indexing barcodes (FC-121-103)(Illumina) and the samples were sequenced on an Illumina HiSeq2500 with 100-bp paired end reads. We sequenced the whole genome, but only mapped the raw sequences to the NC_002084 reference mitogenome sequence.
Data analysis
De-multiplexed raw reads were trimmed using Trimmomatic (Bolger et al. (2014)) version 0.36 and mapped to the mitogenome reference sequence of An. gambiae (Genbank accession number = NC_002084 (Beard et al. (1993))). Freebayes (v1.0.1) (Garrison & Marth, 2013) was used for mitochondrial variant calling assuming single ploidy and without population prior. Mapping statistics were calculated using qualimap version 2.2 (Okonechnikov et al. (2016)) and the data is represented in Table 2. Following the recommendation of Crawford and Lazarro (Crawford & Lazzaro, 2012), we used a minimum depth of 8 to call variants for each individual. Between positions 1-13,470bp of the mitogenome, we obtained consistently high quality reads for all samples, which were used for further analysis. An AT-rich region located between 13,471 and 15,388 suffers from low or zero coverage for sequences generated with the Nextera library preparation kit. Therefore, we excluded these regions from further analysis. The Vcf2fasta program (Danecek et al. (2011)) was used to extract mitogenome sequences from vcf file to fasta format. Geneious version 10.1.3 was used for mitogenome alignments. The phylogenetic tree was generated using PhyloBayes MPI (Lartillot et al., 2013) using the CAT-GTR model on the genomic sequences, which is shown to give similar results compared to amino acid sequences (Foster et al., 2017). We ran the program twice for over 30000 iterations. Max difference between the two runs was 0.045 and minimum effective size was > 100 and created a consensus tree that we visualized in Geneious version 10.1.3. We used scikit-allel (v1.1.9), a software package for Python (Miles & Harding (2017)), to identify species specific markers.
Table 2. List of samples that are used for the study.
Mapped reads indicates the reads that are mapped to the reference genome. Mean coverage indicates the average depth of reads on the mitochondrial DNA and standard deviation indicates the coverage deviation across the mitochondrial DNA.
| Species | Banked_id | Year | Country | Village | Mapped bases | Mean
coverage | Standard
deviation |
|---|
| An. coluzzii-Forest | 11MUTE470 | 2011 | Cameroon | Mutengene | 4265836 | 277.7 | 144.5 |
| An. coluzzii-Forest | 11MUTE472 | 2011 | Cameroon | Mutengene | 1862892 | 121.3 | 23 |
| An. coluzzii-Forest | 11MUTE476 | 2011 | Cameroon | Mutengene | 2130531 | 138.7 | 50.5 |
| An. coluzzii-Forest | 11MUTE477 | 2011 | Cameroon | Mutengene | 806611 | 52.5 | 16.7 |
| An. coluzzii-Forest | 11MUTE480 | 2011 | Cameroon | Mutengene | 804015 | 52.3 | 21 |
| An. coluzzii-Forest | 11MUTE483 | 2011 | Cameroon | Mutengene | 1702247 | 110.8 | 42.9 |
| An. coluzzii-Forest | 11MUTE487 | 2011 | Cameroon | Mutengene | 812839 | 52.9 | 21.2 |
| An. coluzzii-Forest | 11MUTE490 | 2011 | Cameroon | Mutengene | 1882088 | 122.5 | 52.4 |
| An. coluzzii-Forest | 11MUTE491 | 2011 | Cameroon | Mutengene | 1422997 | 92.6 | 46.6 |
| An. coluzzii-Forest | 11MUTE493 | 2011 | Cameroon | Mutengene | 627590 | 40.9 | 17.3 |
| An. coluzzii-Mopti | 12KELA022 | 2012 | Mali | Kela | 3695920 | 240.6 | 64.4 |
| An. coluzzii-Mopti | 12KELA024 | 2012 | Mali | Kela | 574282 | 37.4 | 30.8 |
| An. coluzzii-Mopti | 12KELA046 | 2012 | Mali | Kela | 4152520 | 270.3 | 87.2 |
| An. coluzzii-Mopti | 12KELA085 | 2012 | Mali | Kela | 10883282 | 708.4 | 345 |
| An. coluzzii-Mopti | 12KELA087 | 2012 | Mali | Kela | 3351158 | 218.1 | 79.8 |
| An. coluzzii-Mopti | 12KELA088 | 2012 | Mali | Kela | 1704283 | 110.9 | 91.3 |
| An. coluzzii-Mopti | 12KELA099 | 2012 | Mali | Kela | 349531 | 22.8 | 11 |
| An. coluzzii-Mopti | 12KELA112 | 2012 | Mali | Kela | 8550102 | 556.5 | 198.2 |
| An. coluzzii-Mopti | 12KELA161 | 2012 | Mali | Kela | 33794208 | 2199.7 | 629.3 |
| An. gambiae-Savannah | 12KELA210 | 2012 | Mali | Kela | 3007375 | 195.8 | 53.3 |
| An. gambiae-Bamako | 12KELA214 | 2012 | Mali | Kela | 26441050 | 1721.1 | 566.4 |
| An. gambiae-Bamako | 12KELA219 | 2012 | Mali | Kela | 3617355 | 235.5 | 130.2 |
| An. gambiae-Savannah | 12KELA228 | 2012 | Mali | Kela | 7783776 | 506.7 | 262.8 |
| An. gambiae-Savannah | 12KELA233 | 2012 | Mali | Kela | 7827363 | 509.5 | 138.6 |
| An. gambiae-Savannah | 12KELA234 | 2012 | Mali | Kela | 6721204 | 437.5 | 205.9 |
| An. gambiae-Bamako | 12KELA239 | 2012 | Mali | Kela | 6683521 | 435 | 126.4 |
| An. gambiae-Bamako | 12KELA240 | 2012 | Mali | Kela | 15131480 | 984.9 | 270.8 |
| An. gambiae-Bamako | 12KELA244 | 2012 | Mali | Kela | 12851754 | 836.5 | 306.5 |
| An. gambiae-Savannah | 12KELA285 | 2012 | Mali | Kela | 407888 | 26.6 | 119.8 |
| An. gambiae-Savannah | 12KELA321 | 2012 | Mali | Kela | 1034014 | 67.3 | 43.8 |
| An. gambiae-Savannah | 12KELA334 | 2012 | Mali | Kela | 20949015 | 1363.6 | 400.4 |
| An. gambiae-Savannah | 12KELA348 | 2012 | Mali | Kela | 12053890 | 784.6 | 280.9 |
| An. gambiae-Bamako | 12KELA367 | 2012 | Mali | Kela | 12109235 | 788.2 | 240.1 |
| An. coluzzii-Mopti | 12KELA400 | 2012 | Mali | Kela | 13707820 | 892.3 | 398.2 |
| An. gambiae-Bamako | 12KELA406 | 2012 | Mali | Kela | 17605437 | 1146 | 463.2 |
| An. gambiae-Savannah | 12KELA409 | 2012 | Mali | Kela | 10526480 | 685.2 | 259.1 |
| An. coluzzii-Mopti | 12KELA420 | 2012 | Mali | Kela | 31785953 | 2069 | 845.5 |
| An. gambiae-Bamako | 12KELA443 | 2012 | Mali | Kela | 25740781 | 1675.5 | 669.1 |
| An. gambiae-Bamako | 12KELA457 | 2012 | Mali | Kela | 1360654 | 88.6 | 36.6 |
| An. coluzzii-Mopti | 12KELA458 | 2012 | Mali | Kela | 153686 | 10 | 10.4 |
| An. gambiae-Bamako | 12KELA467 | 2012 | Mali | Kela | 10499093 | 683.4 | 249.1 |
| An. gambiae-Savannah | 12KELA468 | 2012 | Mali | Kela | 10315033 | 671.4 | 197.1 |
| An. gambiae-Bamako | 12KELA481 | 2012 | Mali | Kela | 20308589 | 1321.9 | 307.6 |
| An. coluzzii-Mopti | 12KELA496 | 2012 | Mali | Kela | 2975297 | 193.7 | 162.9 |
| An. gambiae-Bamako | 12KELA651 | 2012 | Mali | Kela | 376689 | 24.5 | 11.3 |
| An. gambiae-Savannah | 12KELA812 | 2012 | Mali | Kela | 799071 | 52 | 29.3 |
| An. arabiensis | 12LUPI001 | 2012 | Tanzania | Lupiro | 2843317 | 185.1 | 34.9 |
| An. arabiensis | 12LUPI007 | 2012 | Tanzania | Lupiro | 6288802 | 409.3 | 40 |
| An. arabiensis | 12LUPI024 | 2012 | Tanzania | Lupiro | 6328898 | 412 | 78.5 |
| An. arabiensis | 12LUPI056 | 2012 | Tanzania | Lupiro | 5440256 | 354.1 | 39.2 |
| An. arabiensis | 12LUPI059 | 2012 | Tanzania | Lupiro | 39721262 | 2585.5 | 801.8 |
| An. arabiensis | 12LUPI071 | 2012 | Tanzania | Lupiro | 3433158 | 223.5 | 59.2 |
| An. arabiensis | 12LUPI074 | 2012 | Tanzania | Lupiro | 10096062 | 657.2 | 100.5 |
| An. arabiensis | 12LUPI082 | 2012 | Tanzania | Lupiro | 5732773 | 373.2 | 69.6 |
| An. arabiensis | 12MINE001 | 2012 | Tanzania | Minepa | 7768923 | 505.7 | 66.9 |
| An. arabiensis | 12MINE040 | 2012 | Tanzania | Minepa | 2784428 | 181.2 | 54.9 |
| An. arabiensis | 12MINE100 | 2012 | Tanzania | Minepa | 10753877 | 700 | 93.9 |
| An. arabiensis | 12MINE101 | 2012 | Tanzania | Minepa | 5684230 | 370 | 41.9 |
| An. arabiensis | 12MINE105 | 2012 | Tanzania | Minepa | 1526829 | 99.4 | 32.8 |
| An. arabiensis | 12MINE111 | 2012 | Tanzania | Minepa | 5578562 | 363.1 | 76.3 |
| An. arabiensis | 12SAGA066 | 2012 | Tanzania | Sagamaganga | 12745079 | 829.6 | 142.3 |
| An. arabiensis | 12SAGA107 | 2012 | Tanzania | Sagamaganga | 14460217 | 941.2 | 259.2 |
| An. arabiensis | 12SAGA131 | 2012 | Tanzania | Sagamaganga | 15333239 | 998.1 | 282.9 |
| An. arabiensis | 12SAGA133 | 2012 | Tanzania | Sagamaganga | 3792945 | 246.9 | 62.5 |
| An. arabiensis | 12SAGA134 | 2012 | Tanzania | Sagamaganga | 2439101 | 158.8 | 34.5 |
| An. arabiensis | 12SAGA141 | 2012 | Tanzania | Sagamaganga | 3130504 | 203.8 | 33.3 |
| An. arabiensis | 05OKJ017 | 2005 | Cameroon | Ourodoukoudje | 9041052 | 588.5 | 78.8 |
| An. arabiensis | 05OKJ042 | 2005 | Cameroon | Ourodoukoudje | 148752684 | 9682.5 | 785.7 |
| An. arabiensis | 05OKJ045 | 2005 | Cameroon | Ourodoukoudje | 35514980 | 2311.7 | 262.8 |
| An. arabiensis | 05OKJ070 | 2005 | Cameroon | Ourodoukoudje | 22847478 | 1487.2 | 400.5 |
Dataset 1.Aligned FASTA file of mitogenome samples.
Results and Discussion
We identified a total of 783 single nucleotide polymorphisms (SNPs) over the entire mitogenome. The majority of these (58.7%) were singletons (found on one of the 70 mitogenomes). We did not identify any SNPs unique to the species or chromosomal forms (Supplementary Table S1) and therefore conclude that mtDNA is not suitable for Anopheles gambiae complex species identification.
The lack of species-specific markers is also reflected in the phylogenetic tree (Figure 1). An. arabiensis, An. coluzzii and An. gambiae did not cluster separately, which is consistent with previous reports that compared mitochondrial genome sequence data from specimens originating from Kenya, Senegal and South Africa (Besansky et al. (1997)) and Burkina Faso, Cameroon, Kenya, Mali, South Africa, Tanzania and Zimbabwe (Fontaine et al. (2015), Supplemental material).
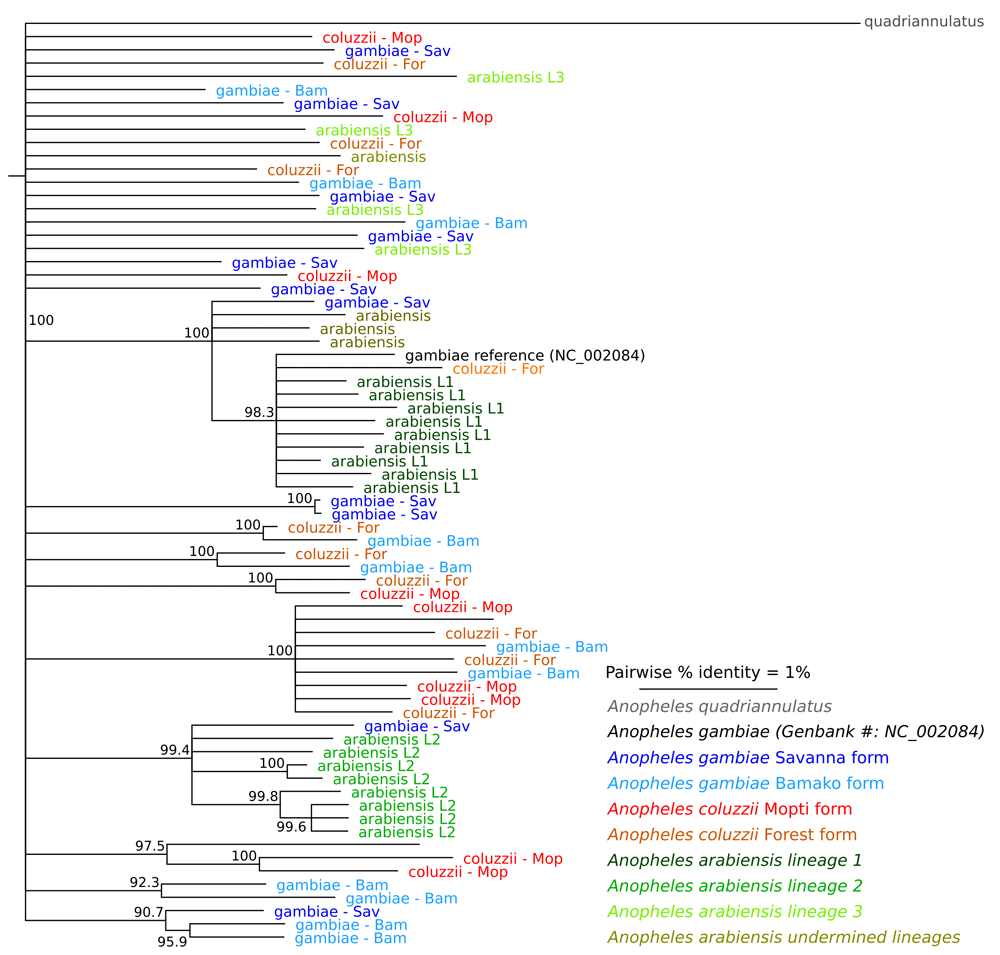
Figure 1. Phylogenetic tree inferred from mtDNA genome sequence data.
The phylogenetic tree fails to reveal a clear division of the operational taxonomic units included in this analysis. Colors indicate the species or chromosomal form and numbers at the branches indicate the accuracy of the inferred branches on a scale of 0–1, where 1 represents the highest confidence. The three An. arabiensis lineages are previously reported by Maliti and co-workers (Maliti et al., 2016).
Our data may indicate that there is no divergent selection in mitogenome among An. gambiae complex. Since mitochondrial genomes have a higher (1–10 times) substitution rate than nuclear genomes (Havird & Sloan, 2016; Lynch & Walsh, 2007), one might expect some level of divergence in the mitogenome in the absence of selection if the taxa have been separated by reproductive barrier even if they are in sympatry just as people have observed in nuclear genome. Therefore, our data showing lack of any species-specific markers on the mitogenome may due to the results of episodic hybridizations occurred between two species. Of note, 36 of the samples that we used in our study originated from Kela (Mali). Kela is located near the village of Selinkenyi, where previous studies have shown a history of hybridization and introgression between An. gambiae and An. coluzzii (Lee et al. (2013); Main et al. (2015); Norris et al. (2015)), which may have resulted in shared polymorphisms in their mitochondrial genomes. Shared polymorphisms in their mitochondrial genomes, where history has not been reported, also appeared to have occurred in Mutengene (Cameroon), where both An. gambiae and An. coluzzii occur sympatrically. Hybridization between either An. coluzzii or An. gambiae with An. arabiensis yields sterile males (Slotman et al. (2004)), but phylogenomic analysis of these species show patterns of introgression between all of them (Fontaine et al. (2015)), which could be the reason that we do not find any species-specific markers on the mitogenome. Our mitochondrial genome study does not provide conclusive evidence for hybridization and introgression among the taxa under study. However, our data suggest that this is a possibility and it would be consistent with results reported by (Fontaine et al., 2015) and (Besansky et al., 1997). Future modeling work may illuminate the likely contribution of different evoluationary forces that shapes mitogenome and nuclear genome evolution.
Data availability
Aligned sequences were submitted to the National Center for Biotechnology Information (NCBI) Accession number: MG930826 - MG930896
Dataset 1. Aligned FASTA file of mitogenome samples 10.5256/f1000research.13807.d192892 (Hanemaaijer et al., 2018)
Grant information
We thank University of California - Irvine, Malaria Initiatives (UCIMI) for their support.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
We thank Michelle Sanford for her assistance in the field collection in Cameroon in 2011. We thank Clare Marsden for providing the raw data of An. arabiensis samples.
Supplementary material
Supplementary Table S1. List of SNP variants in the different Anopheles species and chromosomal forms.
Click here to access the data
Faculty Opinions recommendedReferences
- Beard CB, Hamm DM, Collins FH:
The mitochondrial genome of the mosquito Anopheles gambiae: DNA sequence, genome organization, and comparisons with mitochondrial sequences of other insects.
Insect Mol Biol.
1993; 2(2): 103–124. PubMed Abstract
| Publisher Full Text
- Besansky N, Krzywinski J, Lehmann T, et al.:
Semipermeable species boundaries between Anopheles gambiae and Anopheles arabiensis: evidence from multilocus DNA sequence variation.
Proc Natl Acad Sci U S A.
2003; 100(19): 10818–10823. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Besansky NJ, Lehmann T, Fahey GT, et al.:
Patterns of mitochondrial variation within and between African malaria vectors, Anopheles gambiae and An. arabiensis, suggest extensive gene flow.
Genetics.
1997; 147(4): 1817–1828. PubMed Abstract
| Free Full Text
- Besansky NJ, Lehmann T, Fahey GT, et al.:
Patterns of mitochondrial variation within and between African malaria vectors, Anopheles gambiae and An. arabiensis, suggest extensive gene flow.
Genetics.
1997; 147(4): 1817–1828. PubMed Abstract
| Free Full Text
- Bolger AM, Loshe M, Usadel B:
Trimmomatic: a flexible trimmer for Illumina sequence data.
Bioinformatics.
2014; 30(15): 2114–2120. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Brown WM, George M Jr, Wilson AC:
Rapid evolution of animal mitochondrial DNA.
Proc Natl Acad Sci U S A.
1979; 76(4): 1967–1971. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Coetzee M, Hunt RH, Wilkerson R, et al.:
Anopheles coluzzii and Anopheles amharicus, new members of the Anopheles gambiae complex.
Zootaxa.
2013; 3619(3): 246–274. PubMed Abstract
| Publisher Full Text
- Crawford JE, Lazzaro BP:
Assessing the accuracy and power of population genetic inference from low-pass next-generation sequencing data.
Front Genet.
2012; 3: 66. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Cronin MA, Palmisciano DA, Vyse ER, et al.:
Mitochondrial DNA in wildlife forensic science: species identification of tissues.
Wildlife Soc B (1973-2006).
1991; 19(1): 94–105. Reference Source
- Danecek P, Auton A, Abecasis G, et al.:
The variant call format and VCFtools.
Bioinformatics.
2011; 27(15): 2156–58. PubMed Abstract
| Publisher Full Text
| Free Full Text
- De Barba M, Adams JR, Goldberg CS, et al.:
Molecular species identification for multiple carnivores.
Conserv Genet Resour.
2014; 6(4): 821–824. Publisher Full Text
- De Mandal S, Chhakchhuak L, Gurusubramanian G, et al.:
Mitochondrial markers for identification and phylogenetic studies in insects – A Review.
DNA Barcodes.
2014; 2(1). Publisher Full Text
- Donnelly M, Pinto J, Girod R, et al.:
Revisiting the role of introgression vs shared ancestral polymorphisms as key processes shaping genetic diversity in the recently separated sibling species of the Anopheles gambiae complex.
Heredity (Edinb).
2004; 92(2): 61–8. PubMed Abstract
| Publisher Full Text
- Fontaine MC, Pease JB, Steele A, et al.:
Mosquito genomics. Extensive introgression in a malaria vector species complex revealed by phylogenomics.
Science.
2015; 347(6217): 1258524. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Fontaine MC, Pease JB, Steele A, et al.:
Mosquito genomics. Extensive introgression in a malaria vector species complex revealed by phylogenomics.
Science.
2015; 347(6217): 1258524. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Foster PG, de Oliveira TMP, Bergo ES, et al.:
Phylogeny of Anophelinae using mitochondrial protein coding genes.
R Soc Open Sci.
2017; 4(11): 170758. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Fu Q, Mittnik A, Johnson PLF, et al.:
A revised timescale for human evolution based on ancient mitochondrial genomes.
Curr Biol.
2013; 23(7): 553–559. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Garrison E, Marth G:
Haplotype-based variant detection from short-read sequencing.
arXiv: 1207.3907.
Cornell University Library; 2013. Reference Source
- Green CA:
Cytological maps for the practical identification of females of the three freshwater species of the Anopheles gambiae complex.
Ann Trop Med Parasitol.
1972; 66(1): 143–7. PubMed Abstract
| Publisher Full Text
- Haag-Liautard C, Coffey N, Houle D, et al.:
Direct estimation of the mitochondrial DNA mutation rate in Drosophila melanogaster.
PLoS Biol.
2008; 6(8): e204. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Hanemaaijer MJ, Houston P, Collier TC, et al.:
Dataset 1: Mitochondrial genomes of Anopheles arabiensis, An. gambiae and An.coluzzii show no clear species division.
F1000Research.
2018. http://www.doi.org/10.5256/f1000research.13807.d192892
- Harrison RG:
Animal mitochondrial DNA as a genetic marker in population and evolutionary biology.
Trends Ecol Evol.
1989; 4(1): 6–11. PubMed Abstract
| Publisher Full Text
- Havird JC, Sloan DB:
The Roles of Mutation, Selection, and Expression in Determining Relative Rates of Evolution in Mitochondrial versus Nuclear Genomes.
Mol Biol Evol.
2016; 33(12): 3042–3053. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Lanzaro GC, Lee Y:
Speciation in Anopheles gambiae — The distribution of genetic polymorphism and patterns of reproductive isolation among natural populations.
Anopheles mosquitoes-New insights into malaria vectors.
InTech, 2013. Publisher Full Text
- Lartillot N, Rodrigue N, Stubbs D, et al.:
PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment.
Syst Biol.
2013; 62(4): 611–615. PubMed Abstract
| Publisher Full Text
- Lee Y, Cornel AJ, Meneses CR, et al.:
Ecological and genetic relationships of the Forest-M form among chromosomal and molecular forms of the malaria vector Anopheles gambiae sensu stricto.
Malar J.
2009; 8: 75. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Lee Y, Marsden CD, Nieman C, et al.:
A new multiplex SNP genotyping assay for detecting hybridization and introgression between the M and S molecular forms of Anopheles gambiae.
Mol Ecol Resour.
2014; 14(2): 297–305. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Lee Y, Marsden CD, Nieman C, et al.:
A new multiplex SNP genotyping assay for detecting hybridization and introgression between the M and S molecular forms of Anopheles gambiae.
Mol Ecol Resour.
2014; 14(2): 297–305. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Lee Y, Marsden CD, Norris LC, et al.:
Spatiotemporal dynamics of gene flow and hybrid fitness between the M and S forms of the malaria mosquito, Anopheles gambiae.
Proc Natl Acad Sci U S A.
2013; 110(49): 19854–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Llamas B, Fehren-Schmitz L, Valverde G, et al.:
Ancient mitochondrial DNA provides high-resolution time scale of the peopling of the Americas.
Sci Adv.
2016; 2(4): e1501385. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Lynch M, Walsh B:
The origins of genome architecture. Sinauer Associates; Sunderland, MA. 2007. Reference Source
- Main BJ, Lee Y, Collier TC, et al.:
Complex genome evolution in Anopheles coluzzii associated with increased insecticide usage in Mali.
Mol Ecol.
2015; 24(20): 5145–5157. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Maliti DV, Marsden C, Main B, et al.:
Investigating associations between biting time in the malaria vector Anopheles arabiensis Patton and single nucleotide polymorphisms in circadian clock genes: support for sub-structure among An. arabiensis in the Kilombero valley of Tanzania.
Parasit Vectors.
2016; 9: 109. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Marsden CD, Lee Y, Kreppel K, et al.:
Diversity, differentiation, and linkage disequilibrium: prospects for association mapping in the malaria vector Anopheles arabiensis.
G3 (Bethesda).
2014; 4(1): 121–131. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Miles A, Harding N:
cggh/scikit-allel: v1.1.8 (Version v1.1.8).
Zenodo.
2017. Publisher Full Text
- Norris LC, Main BJ, Lee Y, et al.:
Adaptive introgression in an African malaria mosquito coincident with the increased usage of insecticide-treated bed nets.
Proc Natl Acad Sci U S A.
2015; 112(3): 815–820. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Okonechnikov K, Conesa A, García-Alcalde F:
Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data.
Bioinformatics.
2016; 32(2): 292–294. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Pegg GG, Sinclair B, Briskey L, et al.:
MtDNA barcode identification of fish larvae in the southern Great Barrier Reef–Australia.
Sci Mar.
2006; 70(S2): 7–12. Reference Source
- Phillips MJ, Haouchar D, Pratt RC, et al.:
Inferring kangaroo phylogeny from incongruent nuclear and mitochondrial genes.
PLoS One.
2013; 8(2): e57745. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Pombi M, Caputo B, Simard F, et al.:
Chromosomal plasticity and evolutionary potential in the malaria vector Anopheles gambiae sensu stricto: insights from three decades of rare paracentric inversions.
BMC Evol Biol.
2008; 8(1): 309. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Scott JA, Brogdon WG, Collins FH:
Identification of single specimens of the Anopheles gambiae complex by the polymerase chain reaction.
Am J Trop Med Hyg.
1993; 49(4): 520–529. PubMed Abstract
| Publisher Full Text
- Scott JA, Brogdon WG, Collins FH:
Identification of single specimens of the Anopheles gambiae complex by the polymerase chain reaction.
Am J Trop Med Hyg.
1993; 49(4): 520–529. PubMed Abstract
| Publisher Full Text
- Shaw KL:
Conflict between nuclear and mitochondrial DNA phylogenies of a recent species radiation: what mtDNA reveals and conceals about modes of speciation in Hawaiian crickets.
Proc Natl Acad Sci U S A.
2002; 99(25): 16122–16127. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Slotman M, Della Torre A, Powell JR:
The genetics of inviability and male sterility in hybrids between Anopheles gambiae and An. arabiensis.
Genetics.
2004; 167(1): 275–287. PubMed Abstract
| Publisher Full Text
| Free Full Text
- Sota T, Vogler AP:
Incongruence of mitochondrial and nuclear gene trees in the Carabid beetles Ohomopterus.
Syst Biol.
2001; 50(1): 39–59. PubMed Abstract
| Publisher Full Text
- Tahir HM, Mehwish, Kanwal N, et al.:
Genetic diversity in cytochrome c oxidase I gene of Anopheles mosquitoes.
Mitochondrial DNA A DNA Mapp Seq Anal.
2016; 27(6): 4298–4301. PubMed Abstract
| Publisher Full Text
- Touré Y, Petrarca V, Traore S, et al.:
The distribution and inversion polymorphism of chromosomally recognized taxa of the Anopheles gambiae complex in Mali, West Africa.
Parassitologia.
1998; 40(4): 477–511. PubMed Abstract
- Turner TL, Hahn MW, Nuzhdin SV:
Genomic islands of speciation in Anopheles gambiae.
PLoS Biol.
2005; 3(9): e285. PubMed Abstract
| Publisher Full Text
| Free Full Text
Comments on this article Comments (0)