Keywords
ClinVar, variation, clinical variant, pathogenic, benign, variant interpretation, variant reclassification, pathogenicity
ClinVar, variation, clinical variant, pathogenic, benign, variant interpretation, variant reclassification, pathogenicity
The dbSNP database1 currently contains over 300 million RefSNPs, and dbVar2 adds over 5 million variant regions to the documented plasticity of the human genome. ClinVar3 is small by comparison, documenting the clinical impact of 300,000 variants. This may seem like a far simpler task; however, the substantial impact of these clinical variants on the lives of patients places a heavier burden on the level of evidence gathering required. Add to this the fragmented nature of the evidence—spread out across publications, databases, predictive software analysis and in individual health records—meaning each of these ClinVar records becomes its own meta-data analysis4. ClinVar, ClinGen5, and the American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP)4 have done an excellent job formulating assertion criteria that allows for a comprehensive analysis of all available data, collating them into a standardized classification. While this became the minimum standard upon its inception, there is still a backlog of older assertions with ill-defined criteria or those missing a specification altogether. Many of these would benefit from submitter reclassification based on the more recent standards.
Given the inconsistent amounts of variant data across the genome and the rapid generation of new studies, the significance of variants also changes at an accelerated pace6. Put in statistical terms, the ClinVar clinical significance represents an estimate of the true population significance, and current estimates are based on limited, often private datasets. In many of the instances of discrepancies in assertion, consensus has been achieved simply by sharing evidence previously unavailable to one party7–9. Harrison et al. found that 87.2% of discordant variants were resolved by reassessment and data sharing8. Several initiatives5,10–14 have had success in encouraging the public sharing of datasets and new studies, but for the foreseeable future, private data will continue to be a challenge to achieving consensus. Clinical assertions based on insufficient evidence can persist in public databases and consequently seed misinformation into future interpretations9,15. Recently, in the field of cardiovascular disease, there have been several high-profile instances of cardiovascular variants deemed to be highly pathogenic, yet not segregating with disease9,16,17. This unfortunate outcome is inevitable owing to the aforementioned reasons and illustrates a key issue: the continual need to share and reconcile new information with old data and reclassify clinical assertions as appropriate on a regular basis6,8.
As a clinician or researcher looking to utilize this information, the depth and sophistication of ClinVar presents a daunting learning curve. This is necessary, as ClinVar houses not only assertions, but evidence, literature, and an impressive amount of cross-reference material3. As Yang et al.18 have suggested, this makes the process of evidence interpretation challenging on an individual variant level and the batch processing of variants even more so. ClinVar itself has provided a utile web interface and simplified data structures for programmatic use19. To the same end, other tools have been developed to address both aims: to easily browse variations and compare curations20,21, or import and manipulate flattened ClinVar data for variant analysis22,23. While the browsing tools allow for user-friendly and web-hosted comparison, they do not provide the throughput to analyze large datasets. Conversely, the local database tools allow for deep analysis on large variant sets, but require a significant amount of programming experience and local computational resources to access and operate.
Clinotator is unique in that it provides largescale batch analysis without necessitating a large local computational resource or deep programming knowledge. It can quickly generate simple annotation tables, annotate vcf files or be integrated into annotation pipelines with little overhead. The goals were two-fold: (i) deliver filtered ClinVar information for each variant, focusing on clinical assertions being made about the variant; and (ii) generate several metrics by which the robustness and consistency of the evidence can be gauged for the overall clinical assertion. Clinotator’s quantification of assertion evidence takes into account significance type, submission age and submitter expertise category for a standardized scoring of clinical impact based on the five ACMG/AMP descriptors of Mendelian disorders: Benign (B), Likely Benign (LB), Uncertain Significance (US), Likely Pathogenic (LP) and Pathogenic (P)4.
Our aim is for Clinotator to be useful in a number of capacities, including prioritizing variants that need reclassification, guiding submitter reconciliation efforts or simply identifying discordant variants for future research targets. To demonstrate its utility, we examined test sets of three-star variants (per ClinVar’s review status star ratings) and variants in ClinVar with “Conflicting Interpretations of pathogenicity” (CI). Clinotator was able to confirm recently published concordance trends6,8,18, and identify several groups of discordant variants for further investigation. It accomplished this efficiently, using a large-scale systematic approach with a minimal computational effort.
Metric calculation. Clinotator collects a variety of characteristics from ClinVar and generates four additional metrics (Table 1). The core component of these metrics is the Clinotator raw score (CTRS), generated as the sum of a variant’s weighted individual clinical assertions (i):
The assertion weight factor (xi) was chosen such that a certain multiple of the next lowest priority significance would be less than or equal to the value of the current significance. The distance between US and LB was then a multiple of 10, and the value of B was a multiple of two higher than LB. No multiple of US could attain LP, which was set as the equivalent positive value to LB; and P was a multiple of two higher than LP. The age of the assertion factor (di) reduces the assertion weight over time after a buffer. For the first 2 years, there is no penalty, then there is a 10% reduction gradation in weight per year through 6 years, at which point the penalty stays at a static 50% reduction thereafter. The submitter class factor (si) is weighted based on ClinVar submitter category as curated by ClinGen, with regular clinical assertions by genetic testing laboratories unweighted at 1.00, expert reviewers receiving a 1.10 and practice guidelines receiving a score of 1.25. Literature-only submissions are filtered out and submissions without assertion criteria or with incomplete data (for example no assertion date provided) are also omitted.
The Clinotator average assertion age (CTAA) is the mean age (in years) of valid clinical assertions. Variant submissions without an assertion age are omitted.
The Clinotator predicted significance (CTPS) is a predicted clinical significance based on the CTRS scores of variants in ClinVar with two or more valid clinical assertions. A dataset of all variants that score two stars in ClinVar and have a Mendelian significance was used as a calibration for the category ranges. For the purposes of this calibration, variants with a Pathogenic/Likely pathogenic (PLP) or Benign/Likely benign (BLB) overall significance were excluded as they could not definitively be placed in either category. Assertions with incomplete assertion data were also excluded. Finally, two-star variants with fewer than two clinical assertions with assertion criteria were excluded. Using this filtered calibration dataset, the bounded regions for each CTPS category were set based on a combination of ClinVar star criteria and non-parametric prediction intervals (PI). The lower bound of each range was set at 2[(assertion weight) * (0.7) * (1.0)]; namely, the minimum ClinVar qualification for two stars with both assertions being no more than 5 years old. The quantiles of each distribution as well as the PIs were examined for a range of confidences. The PI for each clinical significance was chosen as the highest possible confidence that aligned to the above established lower bound. Calculations were conducted in R24, and the non-parametric PIs were defined as the cth and rth values in each distribution, where25,26:
The Clinotator reclassification recommendation (CTRR) is a ranked reclassification priority based on the absolute difference between the ClinVar clinical significance (CVCS) and the CTPS. This field uses the seven values of clinical significance associated with Mendelian diseases (B, BLB, LB, US/CI, LP, PLP, P), valued one through seven. For the purposes of reclassification, CI is scored the same as US. Each shift along the scale increases the rank by one, and a transition between overall zones (all benign ⇔ US/CI ⇔ all pathogenic) adds an additional point.
The total number of points is capped at three. Rankings range from zero to three, in escalating degree of inconsistency.
Software structure. The functional components of Clinotator are contained in four modules and a global variables file. The main program, clinotator.py, handles the I/O, errors and options for various file types.
The getncbi.py module handles querying of the E-utilities database servers27. It splits the input list into batches if necessary (default eLink batch size of 1000) and posts to the Entrez history server. It then fetches xml records in batches (default eFetch batch size of 4500). It handles some minor connection interruptions and gives three retries per batch before giving up. Returned batches are added to a list of xml objects to be handled by variation.py.
The variation.py module defines the VariationClass object, and its methods parse ClinVar xml records and calculate the scoring metrics, which are then stored as instance variables. clinotator.py then utilizes pandas to collect and organize tabled data for output. As the ClinVar xml format is highly sophisticated, it does not frequently lend itself to flattening without considerable database structure. The construction of variation.py will allow for future modification, and storage of additional ClinVar xml data as class attributes, allowing for significant backend manipulation with a minimal footprint on the local machine.
The vcf.py module is dedicated to the handling of vcf as an input type. It stores the header and adds the new INFO field definitions for the new annotation in the output file. The rsIDs in the ID column of the vcf are then sent through the rsID input method. After the annotation table has been created in clinotator.py, vcf.py matches annotations to vcf calls by rsID and alt allele combination. Alt alleles are handled as lists (and ClinVar haplotypes are handled as list instance objects), so multi-allelic loci are correctly labeled with their appropriate ClinVar report. Haplotypes are identified as such, but the ‘vcf_match’ field (Table 1) is omitted from the vcf annotation. The other 12 fields are added to the INFO field as outlined in the vcf version 4.3 standards28.
The global_vars.py file supplies a location for most static variables in the program, including several dictionaries of calibration values. Most of these values do not need any modification, but can be; for instance, download batch sizes from NCBI. If the default values result in frequent http errors, the batch size can be reduced. The maximum eLink batch size (for rsID and vcf types) is 1000 ids, while the maximum eFetch batch size is theoretically 10,000 ids. Both are set to lower levels to reduce the incidence of http errors and can be throttled based on available bandwidth.
Clinotator was designed in a Linux environment and implemented in Python (2.7 or ≥3.4)29, and can run in similar OSX and Windows Python environments. The required modules are pandas (0.20.0 minimum, 0.22.0 recommended)30 and biopython (1.70)31. It can be run on a personal computer with relatively modest system requirements; a minimum of 2 GB available RAM. The command line interface requires three pieces of information: (i) the type of input file, (ii) the file itself, and (iii) your email address. The input file can be one of three types: an rsID list using dbSNP identifiers; a Variation ID (VID) list using ClinVar identifiers; or a vcf file. In each case, multiple files can be included and will be processed in batches. If using a list type file, it should be a plain text file with a list of identifiers, one per line. The email is required by NCBI/biopython.
Additionally, the user can specify several options. A highly recommended log option (--log) generates a text file with the warnings from the run. A more extensive long log file (--long-log) can be specified for annotation details. Both log files override the terminal annotation warnings that occur when Clinotator finds missing xml data in ClinVar records in the default (no log) mode. The log files are written in append mode, so batch runs or multiple runs of Clinotator in the same folder can generate a significantly large log file. Users can also specify the output file prefix (the default is “clinotator”), which will label the output “tsv”, “anno.vcf”, and log files.
In all cases, a tab-delimited table file will be produced. The columns will be the VID, RSID and the fields in Table 1. If a vcf file is selected, Clinotator will generate an additional annotated output vcf file. Annotations are concatenated in the INFO field, including the VID and Table 1. Multi-allelic input variants will include comma-separated values for each minor allele. For further details about installation and usage, see the github repository for this project (Data and software availability section).
All VID lists used in analysis were generated at ClinVar, using the search filters and downloading a UI list in text format. The set of all variants with at least two stars was generated February 24th, 2018, and the set of all CI variants was generated on February 27th, 2018. Both sets of variants were analyzed with Clinotator, and split into two-star, three-star, four-star and CI sets. Additional computational analysis was done using dplyr, ggplot2, ggExtra, gridExtra, and RColorBrewer R packages32–36.
A test set of 10,000 VIDs, was run on a system with a single core from an i7-4770 CPU with 16 GB of available memory. Clinotator averaged 1.79 min to complete, 87% of which comprised the NCBI query and download time. The greatest limitation to run time is the bandwidth of the connection to the NCBI databases. When running the list of all variants with at least two stars in ClinVar (>50,000), the run time never exceeded 15 min, with a post-download parsing time of around 90 s. As Clinotator keeps the NCBI xml results in memory, there can be a substantial memory usage. At the time of writing, the entire ClinVar xml set is approaching 6 GB. Loading the entire set into memory is doable with at least 8 GB of memory, though it is recommended that you batch your queries in this rare case. More typical usage for subsets of ClinVar or batch vcf annotations should not pose a memory issue.
Batch annotation of vcf files is similarly efficient, working on single or multi-sample vcfs. Given the set of seven multi-sample, exonic vcf files available at the 1000 Genomes project, Clinotator was able to generate a variant table and annotate output vcf files for all seven files (15,171 total rsIDs) in an average of 3.94 min, 68% of which was NCBI query and download time. A potential speed limitation to vcf-based annotation is that NCBI is queried for each input vcf file, resulting in duplicate queries of common variants, but the tradeoff is not having to create a local query storage file that may potentially become very large if hundreds or thousands of vcf files are being analyzed in a pipeline. If higher throughput is required, it may be more efficient to consider a variant database structure which can return a non-redundant list of total database rsIDs and utilize the list rsID method to generate a reference table.
A total of 48,483 variants were identified with two or more stars in ClinVar and at least two clinical assertions, with 23 four-star, 5,743 three-star and 42,717 two-star variants. Grouping by stars, there is a consistent average CTAA and CTNA across the various types, around three assertions per variant, and an average of about 1.5 years old. This points to a general continuity in ClinVar, encouraging for previous reports of concordance between different clinical labs and expert review panels18. The only exception is the outlier case of the four-star variant set, which averages about six assertions and an average assertion age of almost four. These variants are a particular group of well-documented CFTR variants, though the practice guideline assertion has not been reevaluated since 2004.
The two-star variants are graphed by CTRS in Figure 1A. The distributions of CTRS widely overlap and significantly skew towards overlarge outliers. The US group is the exception, with a leptokurtic distribution. Notably, despite the weighting of B and P assertion types by twice as much as their “likely” counterparts, the distributions of variants of each zone remain resolutely overlapped. The BLB distribution in particular seems both the largest and the most far ranging, extending beyond the B group. While the P group is slightly more distributed above its family members, the LP and PLP distributions, the PLP distribution still spreads over almost the entire positive side of the spectrum. As the PLP or BLB rating in ClinVar is based on a single piece of each type of evidence, there is not a quantification of how much P/B and how much LP/LB evidence is factored into each assessment.
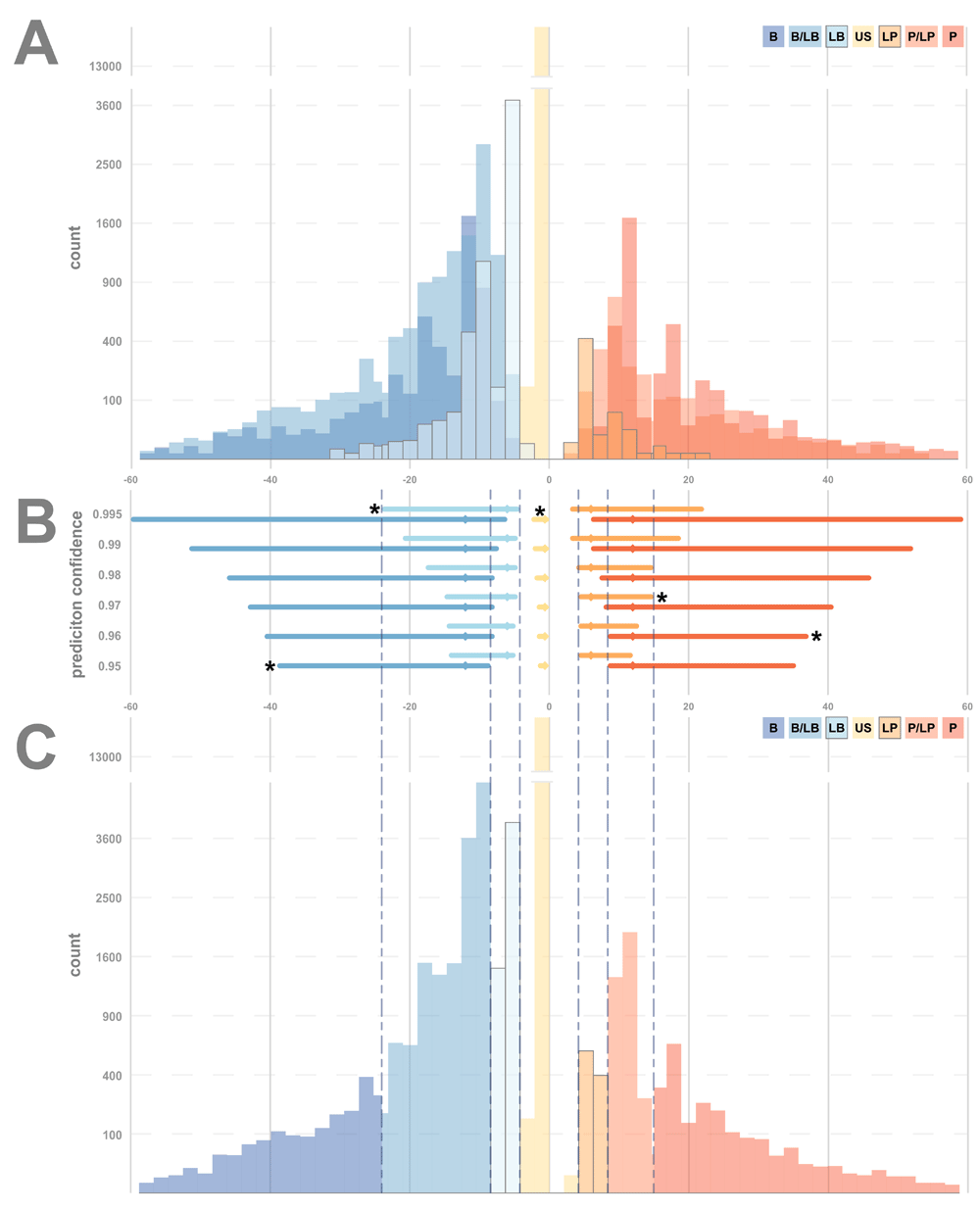
(A) All two-star variants, plotted according to CTRS, and colored based on the seven ClinVar clinical significance designations: Benign (B), Benign/Likely benign (BLB), Likely benign (LB), Uncertain Significance (US), Likely pathogenic (LP) Pathogenic/Likely pathogenic (PLP) and Pathogenic (P). (B) Prediction intervals for the five primary Mendelian clinical significances (B, LB, US, LP and P). Intervals plotted by CTRS value, using five different interval confidences (vertical axis). The optimal confidence interval for each clinical significance is marked with an asterisk. (C) All two-star variants plotted according to Clinotator Raw Score, and colored based on the seven Clinotator predicted significance ranges (B, BLB, LB, US, LP, PLP and P) after calibration with prediction intervals. Dashed lines denote prediction interval boundaries from (B).
A total of 29,252 variants were two-star variants that qualified to be in the five control groups (Table 2). These variants were used to calculate the five prediction intervals depicted in Figure 1B. For each range, the quantiles and prediction intervals were chosen as described above. Given the fixed lower bounds defined by two-star status in ClinVar, the confidence of every prediction interval exceeds the similarly bounded median-centered quantile range, excepting the US category (the center 99.8% of the US distribution is larger than the 99.5% confidence prediction interval). As the US category has no lower bound, its upper bounds are defined by the lower bounds of LB and LP categories, which are outside the entire US control distribution, resulting in a prediction interval confidence greater than 99.99%, still not covering the full width between LB and LP. The likelihood of a US variant falling outside the chosen range is small.
CTRR, Clinotator Reclassification Recommendation. B, Benign; LB, Likely Benign; US, Uncertain Significance; LP, Likely Pathogenic; P, Pathogenic; PI, prediction interval.
The resulting CTPS intervals are shown in Figure 1C, with the BLB and PLP intervals defined by the overlapping prediction intervals. It is worth noting that the overlap between B and LB is much wider than that between P and LP, which reflects the greater overlap of control B and LB distributions. This overlap disparity is observable for all fixed PI confidences individually and in the mixed-confidence PI model used in the final calibration (Figure 1B). Defining BLB and PLP groups with this approach has the advantage of classifying the BLB and PLP quantitatively in a range that cannot be called as either classification by the given confidence, with both classifications exceeding 95% confidence. For this purpose, the overlapping regions of PLP and BLB exist—not as yet another classification bin—as a measure of plasticity of borderline assertions.
A potential concern for non-parametric prediction intervals is that they are inaccurate for values outside the control distribution. However, as the lower bounds are not defined by the prediction interval, only the upper bounds are vulnerable to extreme outliers. Regardless of how far outside the upper interval boundary a given variant may fall, the CTPS determination remains the same, limiting the potential for outliers to impact the reclassification score.
Reclassification recommendations for all of the two-star variants in Figure 2 largely confirm most variants shift by only a single position, if at all (see also Table 2). Only 16 variants are reclassified from an LB, LP, or PLP to a US classification. Most shifts occurred between the overlap categories and their immediate neighbors, likely resulting from the altered definition of the overlap category. Both of these results support previous research showing a fairly high general concordance in ClinVar18,37,38.
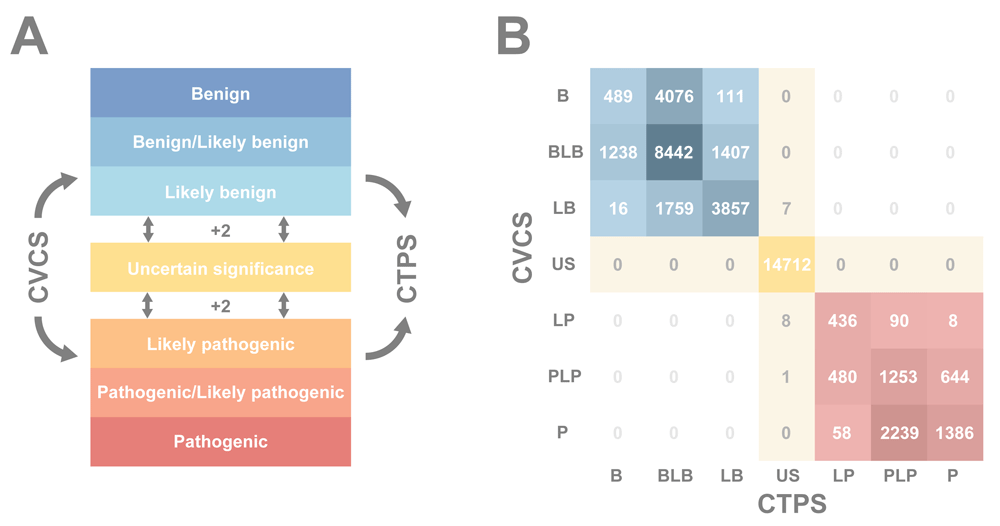
(A) A schematic of the CTRR scoring workflow. ClinVar Clinical Significance (CVCS) is used as a starting point, and each rectangle passed to arrive at the Clinotator Predicted Significance (CTPS) counts as a point. Transitioning a significance family boundary adds an extra point (i.e., moving from Uncertain Significance (US) to Benign/Likely benign (BLB) scores a CTRR = 2 + 1 = 3). (B) A heat map of variant counts with each CVCS and CTPS combination. Darker squares correspond to higher numbers of variants. Blue represents Benign (B), BLB and Likely benign (LB); yellow represents US and red represents Likely pathogenic (LP), Pathogenic/Likely pathogenic (PLP) and Pathogenic (P).
Of variants with at least two stars, all but one of the high priority for reclassification (CTRR = 3) were in the three-star group, and were further investigated. All 56 of these three-star, CTRR rank three variants were of the US classification. As Figure 3A suggests, these variants are primarily predicted to be in the benign family (41 BLB, 1 B). In total, 14 are predicted to be medically significant, belonging to the pathogenic family (9 PLP, 5 P). The submitters with assertion criteria for these 14 are examined in greater detail in Table 3. In 10/14 cases, the expert assertion is the oldest, eight of which are approaching 5 years of age. Additionally, there is a high level of consensus among the three most represented clinical laboratories, with at least two asserting a P or LP in 12/14. It is also notable that 13 of the variants are associated with cancer (Variation ID 42965 is associated with hypertrophic cardiomyopathy). Yang et al. previously found similar trends in clinical lab concordance, age-related discordance and highest concordance among hereditary cancer genes18. As the expert review overrides the other reviews, the tiered system likely disadvantages these variants, making them ideal candidates for reclassification. The full list of 57 variants with a CTRR score of three is available in Supplementary Table 1.
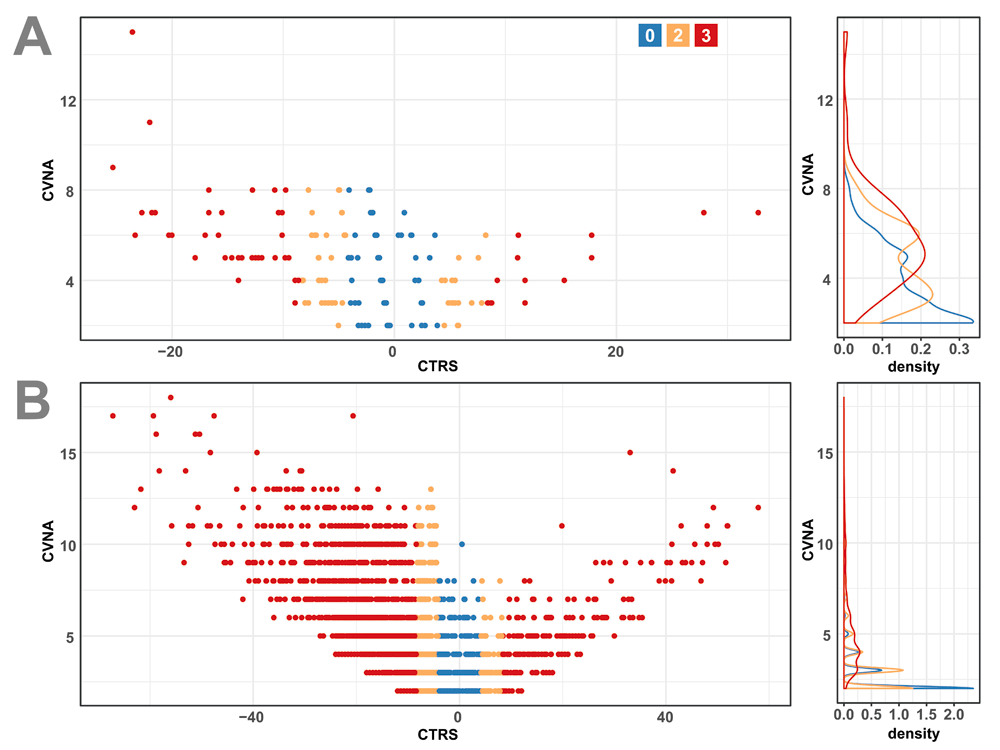
(A) All three-star uncertain significance variants plotted on the left by CVNA and CTRS. On the right is a matched frequency distribution of CVNA. Values are colored according to their Clinotator reclassification recommendation (CTRR): blue represents a score of zero, yellow a score of two and red a score of three. (B) All conflicting interpretations of pathogenicity variants, plotted in the same manner and coloring scheme as (A).
CTPS, Clinotator predicted significance; CTRS, Clinotator raw score; CVNA, ClinVar number of clinical associations; CTAA, average clinical assertion age; LB, Likely Benign; US, Uncertain Significance; LP, Likely Pathogenic; P, Pathogenic; PI, prediction interval.
One-star variants with CI status comprise a set of 13,880 variants. These variants are shown in Figure 3B, and show a similar trend to the Figure 3A distribution of three-star US variants: primarily benign, and increasing CTRR with a larger number of assertions. Looking at the distribution of CI variants with a CTRR of three (Figure 4), there are a number of potential reclassifications, which is unsurprising given their conflicted status. To sample what constitutes a minimum amount of evidence for a CTRR of three, the medically significant variants with only two criteria-based clinical assertions are provided in Table 4 (14 variants of PLP significance). Unlike the variants in Table 3, the majority of these variants are not associated with cancer. Instead, they are associated with cardiovascular diseases, metabolic diseases, and Rett syndrome. Despite not being cancer-focused, there is still a fair amount of concordance among clinical lab assertions. In most cases, the reason for conflict is a single significance provided without assertion criteria, substantially older than the two valid assertions. Given the ages of the conflicting assertions, and the lack of assertion criteria, inviting the submitters to re-evaluate their submissions would most likely reconcile the discrepancies. The full set of CI variants with a CTRR of three are available in Supplementary Table 2.
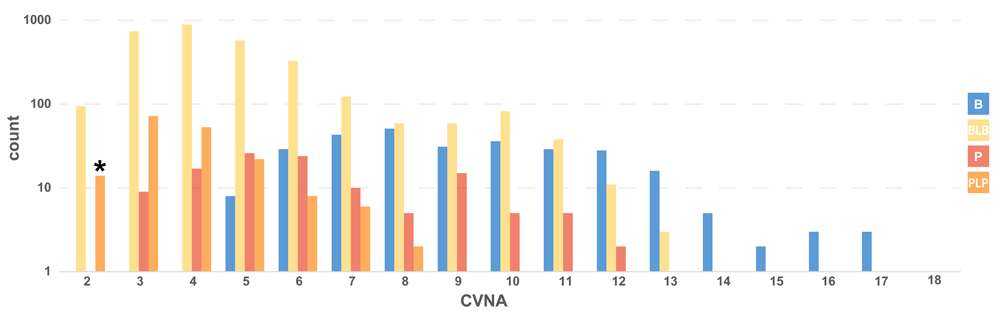
CI variants with a Clinotator reclassification recommendation (CTRR) of three, counted by CVNA and colored by Clinotator predicted significance (CTPS). Blue represents Benign (B), yellow represents Benign/Likely benign (BLB), orange represents Pathogenic/Likely pathogenic (PLP) and red is Pathogenic (P). The asterisk denotes the column of 14 variants examined in Table 4.
B, Benign; LB, Likely Benign; US, Uncertain Significance; LP, Likely Pathogenic; P, Pathogenic; PLP, Pathogenic/Likely pathogenic.
| VID | RSID | CTPS | CTRS | CTAA | date | clinsig | date | clinsig | date | clinsig | date | clinsig |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 12348 | 121912652 | PLP | 12 | 1 | 5/23/2016 | P | 9/23/2016 | P | - | US | 11/30/1990 | P |
| 25269 | 111033768 | PLP | 8.4 | 1.5 | 9/29/2015 | P | 4/7/2016 | LP | 12/4/2012 | B | ||
| 38182 | 81002813 | PLP | 8.4 | 1.5 | 10/2/2015 | P | 3/21/2016 | LP | 5/1/2012 | P | 12/23/2003 | US |
| 53063 | 199473394 | PLP | 10.2 | 2 | 4/14/2017 | P | 1/1/2014 | P | 6/1/2014 | LB | - | - |
| 54153 | 80357164 | PLP | 8.4 | 1.5 | 10/2/2015 | P | 10/17/2016 | LP | 1/31/2014 | US | ||
| 143603 | 61748420 | PLP | 9.6 | 2.5 | 2/8/2013 | P | 6/13/2017 | P | 2/15/2011 | US | ||
| 143738 | 61749723 | PLP | 8.7 | 1.5 | 11/16/2016 | P | 6/30/2015 | LP | 1/21/2008 | US | ||
| 156661 | 587783132 | PLP | 11.4 | 1.5 | 6/15/2016 | P | 9/11/2015 | P | 11/5/2009 | US | ||
| 161516* | 193920774 | PLP | 9 | 1 | 10/7/2016 | LP | 8/3/2016 | P | ||||
| 185705 | 80356913 | PLP | 8.4 | 1 | 6/26/2017 | LP | 10/2/2015 | P | 1/31/2014 | US | ||
| 202509 | 768431507 | PLP | 9 | 0.5 | 8/7/2017 | P | 2/15/2017 | LP | 3/26/2014 | US | ||
| 203805 | 150591260 | PLP | 12 | 0 | 4/24/2017 | P | 5/5/2017 | P | 2/29/2016 | P | 10/31/2017 | US |
| 226353 | 28942078 | PLP | 8.4 | 1.5 | 3/25/2016 | LP | 3/1/2016 | P | 10/4/2013 | US | ||
| 265349 | 183105855 | PLP | 12 | 0.5 | 7/10/2017 | P | 9/14/2016 | P | 9/30/2016 | US | ||
| Clinical assertion, criteria provided | Clinical assertion, no criteria provided OR literature review | |||||||||||
Notably, one of the variants in this list, Variation ID 161516, had a CI significance based on one P, one LP, 18 LP (somatic) and one US (somatic) assertions. The literature has largely not addressed how to reconcile somatic and germline assertions, and the ACMG/AMP guidelines explicitly state they are “not intended for the interpretation of somatic variation”4.
As shown above, Clinotator can be a useful tool for identifying discrepant records amongst a large and complex database. With limited resources, submitters and curators alike can utilize Clinotator metrics for prioritization of reclassifications and research. Additionally, Clinotator can be used to obtain ClinVar information in batch annotations, providing a convenient method to rapidly obtain some simple ClinVar metrics and Clinotator metrics with minimal computational effort. It can be readily integrated into existing pipelines or stand alone as a quick reference.
Clinotator’s ability to identify and filter missing data fields can also be leveraged to clean up older or incomplete submissions in ClinVar. For instance, the list of variations with at least two stars returned over 9000 assertions with a blank ‘Date Last Evaluated’ field, which has become a required field for current submissions. Submitters can check their own assertions to identify their submissions that lack an assertion date.
It should also be noted that Clinotator should not be used as a tool for directly determining clinical significance. Although Clinotator does develop a predicted significance, this is not through the use of primary evidence. The predictive range generated is for rating evidence strength and reclassification impact. Reclassification should always be done using the ACMG/AMP guidelines and assessing all primary evidence available to the researcher.
To compare/analyze variation report quality (a secondary analysis), Clinotator attempts to establish some common criteria. How to combine independent analyses is a particular problem, as these are not individual data points, but professional judgements using a coordinated guideline and overlapping evidence. It has been previously noted that there will always be some level of professional judgement that results in incongruous assertions8, but ultimately this needs to be reconciled to arrive at an overall interpretation by consensus. Mean or median assertion values will not account for the total body of assertions, falling prey to skew or omission, respectively. This is particularly so when there are multiple weighting factors modulating assertion values, thus an aggregated score can better express the total volume of assertions. Clinotator utilizes its raw score, which is an aggregate of these weighted clinical assertions.
A potential issue that arises out of an aggregate model is that lower-level assertions made in a larger volume might artificially inflate the overall value of a variant. For instance, five LP assertions may give a variant the P status, despite no one submitter having enough evidence for the P category. However, while individual assertions utilize overlapping data, each one likely possesses additional private data as evidence. Thus each LP assertion does provide an additive value in terms of overall pathogenicity. We should therefore consider the five ‘Likely pathogenic’ assertions as more likely ‘Pathogenic’. Clinotator highlights this hypothetical five LP variant as a prime candidate for data sharing and reconciliation between the submitters to meet concordance. Clinotator is calibrated on the current, unambiguous two-star data in ClinVar and will be recalibrated on a regular basis to ensure that these boundaries: (i) change with richer information being submitted to ClinVar, and (ii) honor the intent of the ClinVar starring system when possible. In the ideal case, all of the submitters to ClinVar would have all of the data available and the resources to analyse all variants in ClinVar on a regular basis. In such an optimistic context, Clinotator would likely switch to a mean/median model.
Assigning assertion weights to significance types is unfortunately a subjective process. There is not a universal, objective measure of quantity of pathogenicity available in ClinVar, or, arguably, in the literature. In lieu of a more objective metric, a range of assertion weights were tested and the control two-star distributions were examined, as was the set of all two-star variants (42,717 variants; Figure 1). This allowed for the analysis of variants with mixed assertion types, including analysis of CI variants and variants with mixed submitter expertise categories. For the assertion weights we tried, the relative shapes and overlaps of the five control distributions were largely consistent with the final values. Larger assertion weights primarily expanded the tail skew and overall CTRS values, while smaller weights lowered the distance between US and the other classes, shrinking all CTRS values. Expanding the distance between “Likely” and full class members similarly modified overall overlap CTRS values, but the relative overlap trend (that BLB carried a wider range than PLP) did not change. Mixed assertions can never be separated. Ultimately, Clinotator’s assertion weights are relative to class control distributions, so the current values were spread enough to observe comparative differences in overlap, while delineating pathogenicity families with a high degree of confidence. Future versions of Clinotator will need to be periodically recalibrated on current ClinVar distributions, and ultimately may weight assertion types differently if a more objective standard becomes available.
The PI ranges themselves are defined more objectively. As the control distributions are non-normal in several respects, a ranked non-parametric PI is most appropriate26, relying on the fairly large cross sections of the total variant sets in ClinVar (Table 2). Simply setting a static confidence level for the PI would be preferred, but as the lower bound is set by the ClinVar two-star criterion, scaling the whole range is far better than modifying it. As a result, there is a higher confidence in the predictions of some classes than others, but all are at least 95% confident (Figure 1B). As the goal of these prediction ranges is to assess evidential disparities and not to definitively classify variants, having conservatively wide ranges ensures a higher specificity for the CTRR statistic.
The age of the assertion matters. This has been previously identified as an issue6,8,18, but as both the test cases demonstrate, outdated assertions often fail to take into account new evidence and negatively impact classification. One of the key benefits of the current ACMG/AMP criteria is that any assertion must review all previous evidence and existing data available4. Thus while old data never loses its value, old assertions do; particularly if they were made prior to the establishment of the current standards. Reclassification on a regular interval should be a goal for submitters to ClinVar. Counter to the concept of clinical significance as a static value, it is intrinsically dynamic based on the limited availability of evidence. Thus Clinotator weights against the age of assertions after a grace period to ensure that current literature and data are more effectively integrated into the variation report. The maximum threshold of the age weight was set to 0.5 so that a P or B (±6) assertion at or beyond the limit is effectively downgraded to the associated “Likely” category (±3). LB or LP assertions cannot be downgraded to US significance, but are similarly halved in strength.
Finally, the submitter expertise category is a continued confounder in ClinVar. It has become essential for experts in individual conditions to become involved in classification, as different conditions have nuanced profiles of pathogenicity8,9. However, as seen in our test cases, having the expert reviewers supersede all other clinical assertions results in a masking of assertion data. This complication is exacerbated by the age of assertion issue, but more frequent reclassification wouldn’t address the tiered nature of system. Clinotator’s solution is to weight by reviewer status, giving expert reviews a louder voice without drowning out the clinical significance conversation. The specific weights for expertise are subjective, owing to the absence of an objective submission quality metric on which to rank submitter expertise.
Next steps for development involve a variety of fine tuning work. As its metrics are used for analysis, their effectiveness can be assessed and modified, particularly those with subjective elements. An ideal scenario for assertion type weighting would be for submitters to declare the evidence types they utilized, and whether that came from a private resource (i.e. PS4, private data; or PM2, ExAC data). This would allow for assertion type scoring based on an aggregate of evidence without overlap. Similarly, as variant annotations are tracked over time, the submitter expertise category can be calibrated to reflect the total body of experience that a submitter has, or the relative rates of reclassification in the different review status tiers.
The above examples only begin to describe Clinotator’s applications. Clinotator presents a framework for quantitatively assessing ClinVar evidence, and exploration of variants that have unusual Clinotator metrics. Clinotator can also incorporate new utilities to improve its data parsing sophistication, and additional metrics can be included, potentially incorporating new factors such as somatic mutation. Hopefully, it will become a useful tool for curation of ClinVar, and can be integrated with other tools, allowing for the improved classification of variants.
RRID: SCR_016054.
Clinotator source code available from: https://github.com/rbuteriii/clinotator.
Archived source code at the time of publication: https://doi.org/10.5281/zenodo.121020439.
Software license: GNU General Public License v3
Raw data and analysis is available at: https://doi.org/10.5281/zenodo.121027340.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)