Keywords
RNA-seq, workflow, differential transcript usage, Salmon, DRIMSeq, DEXSeq, stageR, tximport
This article is included in the Bioconductor gateway.
This article is included in the RPackage gateway.
RNA-seq, workflow, differential transcript usage, Salmon, DRIMSeq, DEXSeq, stageR, tximport
RNA-seq experiments can be analyzed to detect differences across groups of samples in total gene expression – the total expression produced by all isoforms of a gene – and additionally differences in transcript usage within a gene. If the amount of expression switches among two or more isoforms of a gene, then the total gene expression may not change by a detectable amount, but the differential transcript usage is nevertheless biologically relevant. While many tutorials and workflows in the Bioconductor project address differential gene expression, there are fewer workflows for performing a differential transcript usage analysis, which provides critical and complementary information to a gene-level analysis. Some of the existing Bioconductor packages and functions that can be used to detect differential transcript usage include BitSeq1, DEXSeq (originally designed for differential exon usage)2, diffSpliceDGE from the edgeR package3,4, diffSplice from the limma package5,6, DRIMSeq7, stageR8, and SGSeq9. The Bioconductor package IsoformSwitchAnalyzeR10 is well documented and can be seen as an alternative to this workflow; IsoformSwitchAnalyzeR allows for import of data from various quantification methods, including Salmon, and allows for statistical inference using DRIMSeq, as well as a rank-based statistical test of transcript proportions. In addition, IsoformSwitchAnalyzeR includes functions for obtaining the nucleotide and amino acid sequence consequences of isoform switching, which is not covered in this workflow. Other packages related to splicing can be found at the DifferentialSplicing BiocViews. For more information about the Bioconductor project and its core infrastructure, please refer to the overview by Huber et al.11.
We note that there are numerous other methods for detecting differential transcript usage outside of the Bioconductor project. The DRIMSeq publication is a good reference for these, having descriptions and comparisons with many current methods7. This workflow will build on the methods and vignettes from three Bioconductor packages: DRIMSeq, DEXSeq, and stageR.
Previously, some of the developers of the Bioconductor packages edgeR and DESeq2 have collaborated to develop the tximport package12 for summarizing the output of fast transcript-level quantifiers, such as Salmon13, Sailfish14, and kallisto15. The tximport package focuses on preparing estimated transcript-level counts, abundances and effective transcript lengths, for gene-level statistical analysis using edgeR3, DESeq216 or limma-voom6. tximport produces an offset matrix to accompany gene-level counts, that accounts for a number of RNA-seq biases as well as differences in transcript usage among transcripts of different length that would bias an estimator of gene fold change based on the gene-level counts17. tximport can alternatively produce a matrix of data that is roughly on the scale of counts, by scaling transcript-per-million (TPM) abundances to add up to the total number of mapped reads. This counts-from-abundance approach directly corrects for technical biases and differential transcript usage across samples, obviating the need for the accompanying offset matrix.
Complementary to an analysis of differential gene expression, one can use tximport to import transcript-level estimated counts, and then pass these counts to packages such as DRIMSeq or DEXSeq for statistical analysis of differential transcript usage. Following a transcript-level analysis, one can aggregate evidence of differential transcript usage to the gene level. The stageR package in Bioconductor provides a statistical framework to screen at the gene-level for differential transcript usage with gene-level adjusted p-values, followed by confirmation of which transcripts within the significant genes show differential usage with transcript-level adjusted p-values8. The method controls the overall false discovery rate (OFDR)18 for such a two-stage procedure, which will be discussed in more detail later in the workflow. We believe that stageR represents a principled approach to analyzing transcript usage changes, as the methods can be evaluated against a target error rate in a manner that mimics how the methods will be used in practice. That is, following rejection of the null hypothesis at the gene-level, investigators would likely desire to know which transcripts within a gene participated in the differential usage.
Here we provide a basic workflow for detecting differential transcript usage using Bioconductor packages, following quantification of transcript abundance using the Salmon method. This workflow includes live, runnable code chunks for analysis using DRIMSeq and DEXSeq, as well as for performing stage-wise testing of differential transcript usage using the stageR package. For the workflow, we use data that is simulated, so that we can also evaluate the performance of methods for differential transcript usage, as well as differential gene and transcript expression. The simulation was constructed using distributional parameters estimated from the GEUVADIS project RNA-seq dataset19 quantified by the recount2 project20, including the expression levels of the transcripts, the amount of biological variability of gene expression levels across samples, and realistic coverage of reads along the transcript.
First we describe details of the simulated data, which will be used in the following workflow. Understanding the details of the simulation will be useful for assessing the methods in the later sections. All of the code used to simulate RNA-seq experiments and write paired-end reads to FASTQ files can be found at an associated GitHub repository for the simulation code21, and the reads and quantification files can be downloaded from Zenodo22–25. Salmon13 was used to estimate transcript-level abundances for a single sample (ERR188297) of the GEUVADIS project19, and this was used as a baseline for transcript abundances in the simulation. Transcripts that were associated with estimated counts less than 10 had abundance thresholded to 0, all other transcripts were considered “expressed”. alpine26 was used to estimate realistic fragment GC bias from 12 samples from the GEUVADIS project, all from the same sequencing center (the first 12 samples from CNAG-CRG in Supplementary Table 2 from Love et al.26). DESeq216 was used to estimate mean and dispersion parameters for a Negative Binomial distribution for gene-level counts for 458 GEUVADIS samples provided by the recount2 project20. An example of DESeq2-generated estimates of dispersion per gene can be seen in Supplementary Figure 1. Note that, while gene-level dispersion estimates were used to generate underlying transcript-level counts, additional uncertainty on the transcript-level data is a natural consequence of the simulation, as the transcript-level counts must be estimated (the underlying transcript counts are not provided to the methods).
polyester27 was used to simulate paired-end RNA-seq reads for two groups of 12 samples each, with realistic fragment GC bias, and with dispersion on transcript-level counts drawn from the joint distribution of mean and dispersion values estimated from the GEUVADIS samples. To compare DRIMSeq and DEXSeq in further detail, we generated an additional simulation in which dispersion parameters were assigned to genes via matching on the gene-level count, and then all transcripts of a gene had counts generated using the same per-gene dispersion. The first sample for group 1 and the first sample for group 2 followed the realistic GC bias profile of the same GEUVADIS sample, and so on for all 12 samples. This pairing of the samples was used to generate balanced data, but not used in the statistical analysis. countsimQC28 was used to examine the properties of the simulation relative to the dataset used for parameter estimation, and the full report can be accessed at the associated GitHub repository for simulation code21.
Differential expression across two groups was generated as follows: 70% of the genes were set as null genes, where abundance was not changed across the two groups. For 10% of genes, all isoforms were differentially expressed at a log fold change between 1 and 2.58 (fold change between 2 and 6). The set of transcripts in these genes was classified as DGE (differential gene expression) by construction, and the expressed transcripts were also DTE (differential transcript expression), but they did not count as DTU (differential transcript usage), as the proportions within the gene remained constant. To simulate balanced differential expression, one of the two groups was randomly chosen to be the baseline, and the other group would have its counts multiplied by the fold change. For 10% of genes, a single expressed isoform was differentially expressed at a log fold change between 1 and 2.58. This set of transcripts was DTE by construction. If the chosen transcript was the only expressed isoform of a gene, this counted also as DGE and not as DTU, but if there were other isoforms that were expressed, this counted for both DGE and DTU, as the proportion of expression among the isoforms was affected. For 10% of genes, differential transcript usage was constructed by exchanging the TPM abundance of two expressed isoforms, or, if only one isoform was expressed, exchanging the abundance of the expressed isoform with a non-expressed one. This counted for DTU and DTE, but not for DGE. An MA plot of the simulated transcript abundances for the two groups is shown in Figure 1.
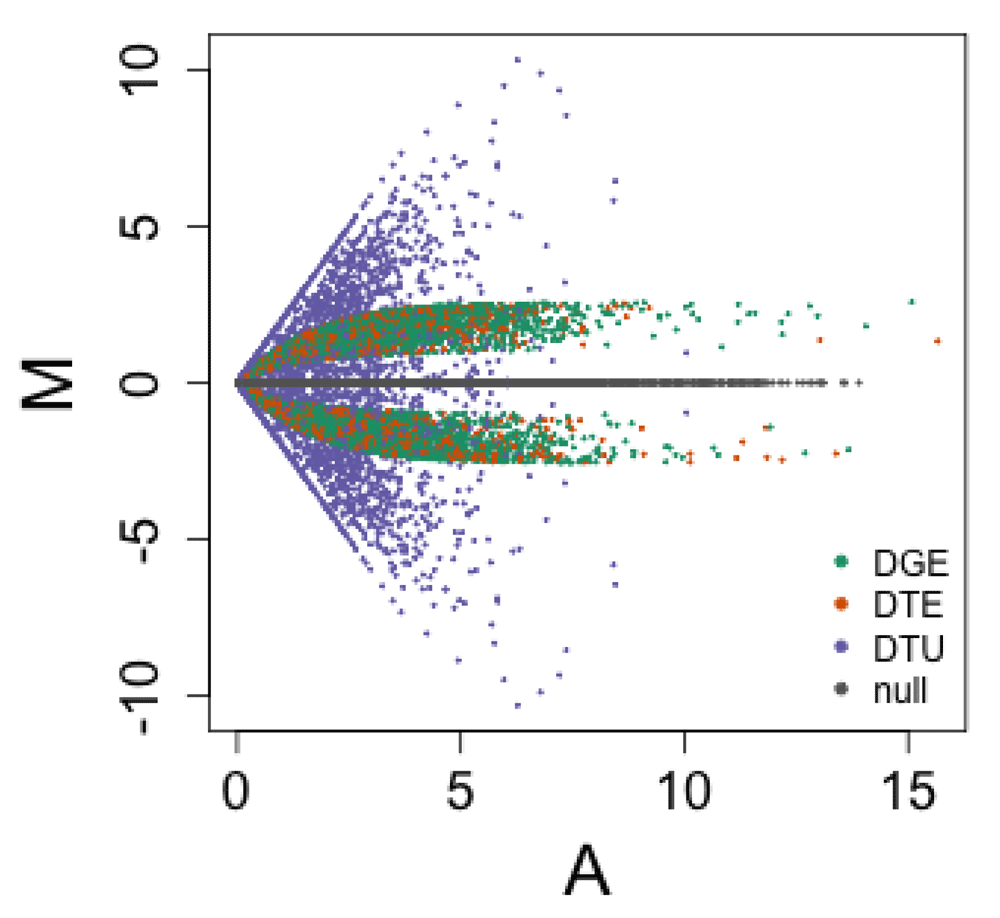
Each point depicts a transcript, with the average log2 abundance in transcripts-per-million (TPM) on the x-axis and the difference between the two groups on the y-axis. Of the transcripts which are expressed with TPM > 1 in at least one group, 77% are null transcripts (grey), which fall by construction on the M=0 line, and 23% are differentially expressed (green, orange, or purple). As transcripts can belong to multiple categories of differential gene expression (DGE), differential transcript expression (DTE), and differential transcript usage (DTU), here the transcripts are colored by which genes they belong to (those selected to be DGE-, DTE-, or DTU-by-construction).
This workflow was designed to work with R 3.5 or higher, and the DRIMSeq, DEXSeq, stageR, and tximport packages for Bioconductor version 3.7 or higher. Bioconductor packages should always be installed following the official instructions. The workflow uses a subset of all genes to speed up the analysis, but the Bioconductor packages can easily be run for this dataset on all human genes on a laptop in less than an hour. Timing for the various packages is included within each section.
We used Salmon version 0.10.0 to quantify abundance and effective transcript lengths for all of the 24 simulated samples. For this workflow, we will use the first six samples from each group. We quantified against the GENCODE human annotation version 28, which was the same reference used to generate the simulated reads. We used the transcript sequences FASTA file that contains “Nucleotide sequences of all transcripts on the reference chromosomes”. When downloading the FASTA file, it is useful to download the corresponding GTF file, as this will be used in later sections.
To build the Salmon index, we used the following command. Recent versions of Salmon will discard identical sequence duplicate transcripts, and keep a log of these within the index directory.
salmon index -t gencode.v28.transcripts.fa -i gencode.v28_salmon-0.10.0To quantify each sample, we used the following command, which says to quantify with six threads using the GENCODE index, with inward and unstranded paired end reads, using fragment GC bias correction, writing out to the directory sample and using as input these two reads files. The library type is specified by -l IU (inward and unstranded) and the options are discussed in the Salmon documentation. Recent versions of Salmon can automatically detect the library type by setting -l A. Such a command can be automated in a bash loop using bash variables, or one can use more advanced workflow management systems such as Snakemake29 or Nextflow30.
salmon quant -p 6 -i gencode.v28_salmon-0.10.0 -l IU \ --gcBias -o sample -1 sample_1.fa.gz -2 sample_2.fa.gz
We can use tximport to import the estimated counts, abundances and effective transcript lengths into R. We recommend to construct a CSV file that keeps track of the sample identifiers and any relevant variables, e.g. condition, time point, batch, and so on. Here we have made a sample CSV file and provided it along with this workflow’s R package.
In order to find this file, we first need to know where on the machine this workflow package lives, so we can point to the extdata directory where the CSV file is located. These two lines of code load the workflow package and find this directory on the machine. These two lines of code would therefore not be part of a typical workflow.
library(rnaseqDTU) csv.dir <- system.file("extdata", package="rnaseqDTU")
The CSV file records which samples are condition 1 and which are condition 2. The columns of this CSV file can have any names, although sample_id will be used later by DRIMSeq, and so using this column name allows us to pass this data.frame directly to DRIMSeq at a later step.
samps <- read.csv(file.path(csv.dir, "samples.csv")) head(samps)
## sample_id condition
## 1 s1_1 1
## 2 s2_1 1
## 3 s3_1 1
## 4 s4_1 1
## 5 s5_1 1
## 6 s6_1 1
samps$condition <- factor(samps$condition) table(samps$condition)
##
## 1 2
## 6 6files <- file.path("/path/to/dir", samps$sample_id, "quant.sf") names(files) <- samps$sample_id head(files)
## s1_1 s2_1
## "/path/to/dir/s1_1/quant.sf" "/path/to/dir/s2_1/quant.sf"
## s3_1 s4_1
## "/path/to/dir/s3_1/quant.sf" "/path/to/dir/s4_1/quant.sf"
## s5_1 s6_1
## "/path/to/dir/s5_1/quant.sf" "/path/to/dir/s6_1/quant.sf"We can then import transcript-level counts using tximport. We suggest for DTU analysis to generate counts from abundance, using the scaledTPM method described by Soneson et al.12. The countsFromAbundance option of tximport uses estimated abundances to generate roughly count-scaled data, such that each column will sum to the number of reads mapped for that library. We recommend scaledTPM for differential transcript usage so that the estimated proportions fit by DRIMSeq in the following sections correspond to the proportions of underlying abundance.
If instead of scaledTPM, we used the original estimated transcript counts (countsFromAbundance="no"), or if we used lengthScaledTPM transcript counts, then a change in transcript usage among transcripts of different length could result in a changed total count for the gene, even if there is no change in total gene expression. This is because the original transcript counts and lengthScaledTPM transcript counts scale with transcript length, while scaledTPM transcript counts do not. For testing DTU using DRIMSeq and DEXSeq, it is convenient if the count-scale data do not scale with transcript length within a gene. Note that this could be corrected by an offset, but this is not easily implemented in the current DTU analysis packages. While this workflow only considers existing software features, we are considering developing a new countsFromAbundance method which would scale abundance for all transcripts of a gene by a fixed gene length, then each sample by its number of mapped reads, therefore balancing between the benefits of scaledTPM and lengthScaledTPM.
The following code chunk is not evaluated, but instead we will load a pre-constructed matrix of counts. The actual quantification files for this dataset have been made publicly available; see the Data availability section at the end of this workflow.
library(tximport) txi <- tximport(files, type="salmon", txOut=TRUE, countsFromAbundance="scaledTPM") cts <- txi$counts cts <- cts[rowSums(cts) > 0,]
Bioconductor offers numerous approaches for building a TxDb object, a transcript database that can be used to link transcripts to genes (among other uses). We ran the following unevaluated code chunks to generate a TxDb, and then used the select function with the TxDb to produce a corresponding data.frame called txdf which links transcript IDs to gene IDs. In this TxDb, the transcript IDs are called TXNAME and the gene IDs are called GENEID. The version 28 human GTF file was downloaded from the GENCODE website when downloading the transcripts FASTA file.
library(GenomicFeatures) gtf <- "gencode.v28.annotation.gtf.gz" txdb.filename <- "gencode.v28.annotation.sqlite" txdb <- makeTxDbFromGFF(gtf) saveDb(txdb, txdb.filename)
Once the TxDb database has been generated and saved, it can be quickly reloaded:
txdb <- loadDb(txdb.filename) txdf <- select(txdb, keys(txdb, "GENEID"), "TXNAME", "GENEID") tab <- table(txdf$GENEID) txdf$ntx <- tab[match(txdf$GENEID, names(tab))]
We load the cts object as created in the tximport code chunks. This contains count-scale data, generated from abundance using the scaledTPM method. The column sums are equal to the number of mapped paired-end reads per experiment. The experiments have between 31 and 38 million paired-end reads that were mapped to the transcriptome using Salmon.
data(salmon_cts) cts[1:3,1:3]
## s1_1 s2_1 s3_1
## ENST00000488147.1 179.798908 184.437348 229.046306
## ENST00000469289.1 0.000000 0.000000 0.000000
## ENST00000466430.5 5.004159 3.627831 9.463167range(colSums(cts)/1e6)
## [1] 31.37738 38.47173We also have the txdf object giving the transcript-to-gene mappings (for construction, see previous section). This is contained in a file called simulate.rda that contains a number of R objects with information about the simulation, that we will use later to assess the methods’ performance.
data(simulate) head(txdf)
## GENEID TXNAME ntx
## 1 ENSG00000000003.14 ENST00000612152.4 5
## 2 ENSG00000000003.14 ENST00000373020.8 5
## 3 ENSG00000000003.14 ENST00000614008.4 5
## 4 ENSG00000000003.14 ENST00000496771.5 5
## 5 ENSG00000000003.14 ENST00000494424.1 5
## 6 ENSG00000000005.5 ENST00000373031.4 2all(rownames(cts) %in% txdf$TXNAME)
## [1] TRUEtxdf <- txdf[match(rownames(cts),txdf$TXNAME),] all(rownames(cts) == txdf$TXNAME)
## [1] TRUEIn order to run DRIMSeq, we build a data.frame with the gene ID, the feature (transcript) ID, and then columns for each of the samples:
counts <- data.frame(gene_id=txdf$GENEID, feature_id=txdf$TXNAME, cts)
We can now load the DRIMSeq package and create a dmDSdata object, with our counts and samps data.frames. Typing in the object name and pressing return will give information about the number of genes:
library(DRIMSeq) d <- dmDSdata(counts=counts, samples=samps) d
## An object of class dmDSdata ## with 16612 genes and 12 samples ## * data accessors: counts(), samples()
The dmDSdata object has a number of specific methods. Note that the rows of the object are gene-oriented, so pulling out the first row corresponds to all of the transcripts of the first gene:
methods(class=class(d))
## [1] [ coerce counts dmFilter dmPrecision length ## [7] names plotData show ## see ’?methods’ for accessing help and source code
counts(d[1,])[,1:4]
## gene_id feature_id s1_1 s2_1
## 1 ENSG00000000419.12 ENST00000371588.9 1394.71411 1210.12539
## 2 ENSG00000000419.12 ENST00000466152.5 135.15850 18.20031
## 3 ENSG00000000419.12 ENST00000371582.8 154.77943 35.39425
## 4 ENSG00000000419.12 ENST00000371584.8 42.85733 86.04958
## 5 ENSG00000000419.12 ENST00000413082.1 0.00000 0.00000It will be useful to first filter the object, before running procedures to estimate model parameters. This greatly speeds up the fitting and removes transcripts that may be troublesome for parameter estimation, e.g. estimating the proportion of expression among the transcripts of a gene when the total count is very low. We first define n to be the total number of samples, and n.small to be the sample size of the smallest group. We use all three of the possible filters: for a transcript to be retained in the dataset, we require that (1) it has a count of at least 10 in at least n.small samples, (2) it has a relative abundance proportion of at least 0.1 in at least n.small samples, and (3) the total count of the corresponding gene is at least 10 in all n samples. We used all three possible filters, whereas only the two count filters are used in the DRIMSeq vignette example code.
It is important to consider what types of transcripts may be removed by the filters, and potentially adjust depending on the dataset. If n was large, it would make sense to allow perhaps a few samples to have very low counts, so lowering min_samps_gene_expr to some factor multiple (< 1) of n, and likewise for the first two filters for n.small. The second filter means that if a transcript does not make up more than 10% of the gene’s expression for at least n.small samples, it will be removed. If this proportion seems too high, for example, if very lowly expressed isoforms are of particular interest, then the filter can be omitted or the min_feature_prop lowered. For a concrete example, if a transcript goes from a proportion of 0% in the control group to a proportion of 9% in the treatment group, this would be removed by the above 10% filter. After filtering, this dataset has 7,764 genes.
n <- 12 n.small <- 6 d <- dmFilter(d, min_samps_feature_expr=n.small, min_feature_expr=10, min_samps_feature_prop=n.small, min_feature_prop=0.1, min_samps_gene_expr=n, min_gene_expr=10) d
## An object of class dmDSdata ## with 7764 genes and 12 samples ## * data accessors: counts(), samples()
The dmDSdata object only contains genes that have more that one isoform, which makes sense as we are testing for differential transcript usage. We can find out how many of the remaining genes have N isoforms by tabulating the number of times we see a gene ID, then tabulating the output again:
table(table(counts(d)$gene_id))
##
## 2 3 4 5 6 7
## 4062 2514 931 222 34 1We create a design matrix, using a design formula and the sample information contained in the object, accessed via samples. Here we use a simple design with just two groups, but more complex designs are possible. For some discussion of complex designs, one can refer to the vignettes of the limma, edgeR, or DESeq2 packages.
design_full <- model.matrix(~condition, data=DRIMSeq::samples(d)) colnames(design_full)
## [1] "(Intercept)" "condition2"Only for speeding up running the live code chunks in this workflow, we subset to the first 250 genes, representing about one thirtieth of the dataset. This step would not be run in a typical workflow.
d <- d[1:250,] 7764 / 250
## [1] 31.056We then use the following three functions to estimate the model parameters and test for DTU. We first estimate the precision, which is related to the dispersion in the Dirichlet Multinomial model via the formula below. Because precision is in the denominator of the right hand side of the equation, they are inversely related. Higher dispersion – counts more variable around their expected value – is associated with lower precision. For full details about the DRIMSeq model, one should read both the detailed software vignette and the publication7. After estimating the precision, we fit regression coefficients and perform null hypothesis testing on the coefficient of interest. Because we have a simple two-group model, we test the coefficient associated with the difference between condition 2 and condition 1, called condition2. The following code takes about half a minute, and so a full analysis on this dataset takes about 15 minutes on a laptop.
set.seed(1) system.time({ d <- dmPrecision(d, design=design_full) d <- dmFit(d, design=design_full) d <- dmTest(d, coef="condition2") })
## ! Using a subset of 0.1 genes to estimate common precision !## ! Using common_precision = 21.2862 as prec_init !## ! Using 0 as a shrinkage factor !## user system elapsed
## 34.213 0.450 35.846To build a results table, we run the results function. We can generate a single p-value per gene, which tests whether there is any differential transcript usage within the gene, or a single p-value per transcript, which tests whether the proportions for this transcript changed within the gene:
res <- DRIMSeq::results(d) head(res)
## gene_id lr df pvalue adj_pvalue
## 1 ENSG00000000457.13 1.493561 4 8.277814e-01 9.120246e-01
## 2 ENSG00000000460.16 1.068294 3 7.847330e-01 9.101892e-01
## 3 ENSG00000000938.12 4.366806 2 1.126575e-01 2.750169e-01
## 4 ENSG00000001084.11 1.630085 3 6.525877e-01 8.643316e-01
## 5 ENSG00000001167.14 28.402587 1 9.853354e-08 5.007113e-07
## 6 ENSG00000001461.16 9.815460 1 1.730510e-03 6.732766e-03res.txp <- DRIMSeq::results(d, level="feature") head(res.txp)
## gene_id feature_id lr df pvalue adj_pvalue
## 1 ENSG00000000457.13 ENST00000367771.10 0.16587607 1 0.6838032 0.9171007
## 2 ENSG00000000457.13 ENST00000367770.5 0.01666448 1 0.8972856 0.9788571
## 3 ENSG00000000457.13 ENST00000367772.8 1.02668495 1 0.3109386 0.6667146
## 4 ENSG00000000457.13 ENST00000423670.1 0.06046507 1 0.8057624 0.9323782
## 5 ENSG00000000457.13 ENST00000470238.1 0.28905766 1 0.5908250 0.8713427
## 6 ENSG00000000460.16 ENST00000496973.5 0.83415788 1 0.3610730 0.7232298Because the pvalue column may contain NA values, we use the following function to turn these into 1’s. The NA values would otherwise cause problems for the stage-wise analysis.
no.na <- function(x) ifelse(is.na(x), 1, x) res$pvalue <- no.na(res$pvalue) res.txp$pvalue <- no.na(res.txp$pvalue)
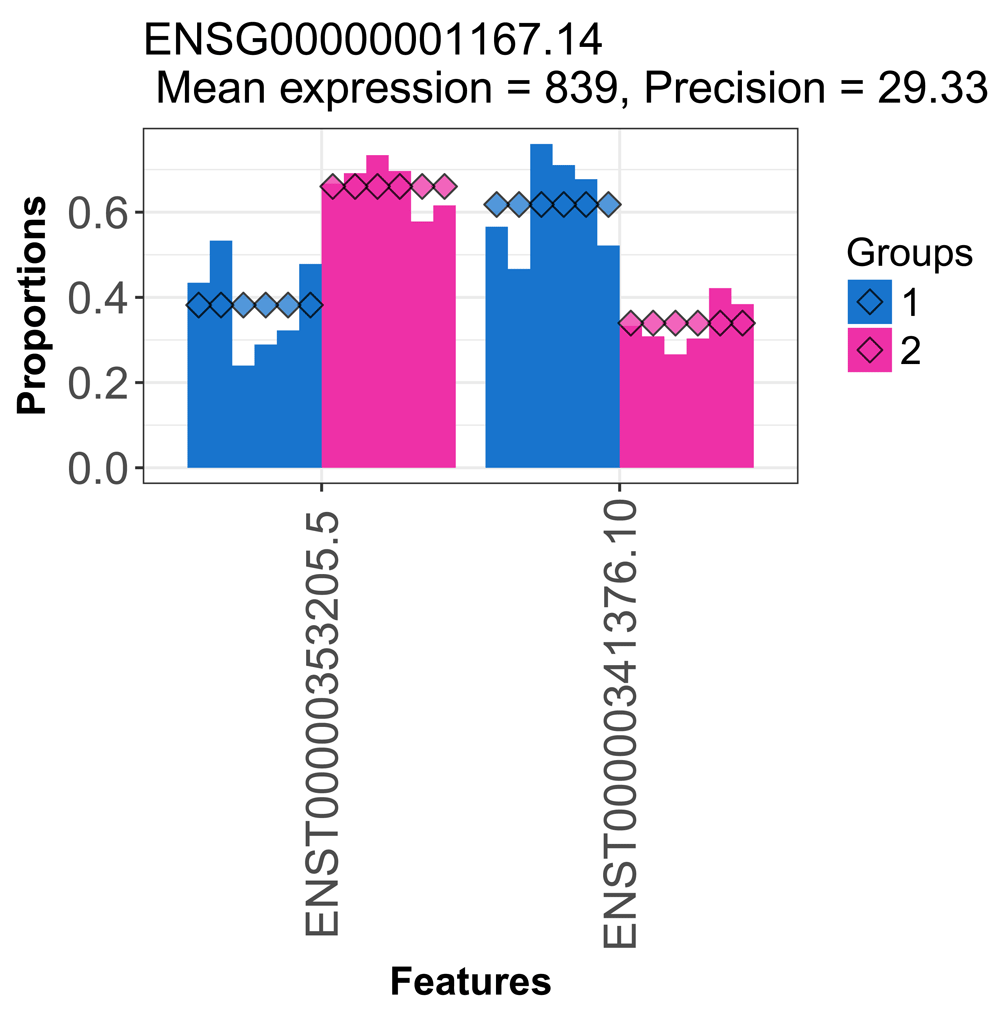
We can plot the estimated proportions for one of the significant genes, where we can see evidence of switching (Figure 2).
idx <- which(res$adj_pvalue < 0.05)[1] res[idx,]
## gene_id lr df pvalue adj_pvalue
## 5 ENSG00000001167.14 28.40259 1 9.853354e-08 5.007113e-07plotProportions(d, res$gene_id[idx], "condition")
Because we have been working with only a subset of the data, we now load the results tables that would have been generated by running DRIMSeq functions on the entire dataset.
data(drim_tables) nrow(res)
## [1] 7764nrow(res.txp)
## [1] 20711A typical analysis of differential transcript usage would involve asking first: “which genes contain any evidence of DTU?”, and secondly, “which transcripts in the genes that contain some evidence may be participating in the DTU?” Note that a gene may pass the first stage without exhibiting enough evidence to identify one or more transcripts that are participating in the DTU. The stageR package is designed to allow for such two-stage testing procedures, where the first stage is called a screening stage and the second stage a confirmation stage8. The methods are general, and can also be applied to testing, for example, changes across a time series followed by investigation of individual time points, as shown in the stageR package vignette. We show below how stageR is used to detect DTU and how to interpret its output.
We first construct a vector of p-values for the screening stage. Because of how the stageR package will combine transcript and gene names, we need to strip the gene and transcript version numbers from their Ensembl IDs (this is done by keeping only the first 15 characters of the gene and transcript IDs).
pScreen <- res$pvalue strp <- function(x) substr(x,1,15) names(pScreen) <- strp(res$gene_id)
We construct a one column matrix of the confirmation p-values:
pConfirmation <- matrix(res.txp$pvalue, ncol=1) rownames(pConfirmation) <- strp(res.txp$feature_id)
We arrange a two column data.frame with the transcript and gene identifiers.
tx2gene <- res.txp[,c("feature_id", "gene_id")] for (i in 1:2) tx2gene[,i] <- strp(tx2gene[,i])
The following functions then perform the stageR analysis. We must specify an alpha, which will be the overall false discovery rate target for the analysis, defined below. Unlike typical adjusted p-values or q-values, we cannot choose an arbitrary threshold later: after specifying alpha=0.05, we need to use 5% as the target in downstream steps. There are also convenience functions getSignificantGenes and getSignificantTx, which are demonstrated in the stageR vignette.
library(stageR) stageRObj <- stageRTx(pScreen=pScreen, pConfirmation=pConfirmation, pScreenAdjusted=FALSE, tx2gene=tx2gene) stageRObj <- stageWiseAdjustment(stageRObj, method="dtu", alpha=0.05) suppressWarnings({ drim.padj <- getAdjustedPValues(stageRObj, order=FALSE, onlySignificantGenes=TRUE) }) head(drim.padj) ## geneID txID gene transcript ## 1 ENSG00000001167 ENST00000341376 1.446731e-05 0.000000 ## 2 ENSG00000001167 ENST00000353205 1.446731e-05 0.000000 ## 3 ENSG00000001461 ENST00000003912 8.263160e-03 0.000000 ## 4 ENSG00000001461 ENST00000339255 8.263160e-03 0.000000 ## 5 ENSG00000001631 ENST00000394507 1.287012e-04 0.060474 ## 6 ENSG00000001631 ENST00000475770 1.287012e-04 1.000000
The final table with adjusted p-values summarizes the information from the two-stage analysis. Only genes that passed the filter are included in the table, so the table already represents screened genes. The transcripts with values in the column, transcript, less than 0.05 pass the confirmation stage on a target 5% overall false discovery rate, or OFDR. This means that, in expectation, no more than 5% of the genes that pass screening will either (1) not contain any DTU, so be falsely screened genes, or (2) contain a transcript with a transcript adjusted p-value less than 0.05 which does not participate in DTU, so contain a falsely confirmed transcript. The stageR procedure allows us to look at both the genes that passed the screening stage and the transcripts with adjusted p-values less than our target alpha, and understand what kind of overall error rate this procedure entails. This cannot be said for an arbitrary procedure of looking at standard gene adjusted p-values and transcript adjusted p-values, where the adjustment was performed independently.
We found that DRIMSeq was sensitive to detect DTU, but could exceed its false discovery rate (FDR) bounds, particularly on the transcript-level tests, and that a post-hoc, non-specific filtering of the DRIMSeq transcript p-values improved the FDR control. We considered the standard deviation (SD) of the per-sample proportions as a filtering statistic. This statistic does not use the information about which samples belong to which condition group. We set the p-values for transcripts with small per-sample proportion SD to 1 and then re-computed the adjusted p-values using the method of Benjamini and Hochberg31. Excluding transcripts with small SD of the per-sample proportions brought the observed FDR closer to its nominal target in the simulation considered here, as shown below.
res.txp.filt <- DRIMSeq::results(d, level="feature") getSampleProportions <- function(d) { cts <- as.matrix(subset(counts(d), select=-c(gene_id, feature_id))) gene.cts <-rowsum(cts, counts(d)$gene_id) total.cts <- gene.cts[match(counts(d)$gene_id), rownames(gene.cts)),] cts/total.cts } prop.d <- getSampleProportions(d) res.txp.filt$prop.sd <- sqrt(rowVars(prop.d)) res.txp.filt$pvalue[res.txp.filt$prop.sd < .1] <- 1 res.txp.filt$adj_pvalue <- p.adjust(res.txp.filt$pvalue, method="BH")
The above post-hoc filter is not part of the DRIMSeq modeling steps, and to avoid interfering with the modeling, we run it after DRIMSeq. The other three filters used before have been tested by the DRIMSeq package authors, and are therefore a recommended part of an analysis before the modeling begins.
The DEXSeq package was originally designed for detecting differential exon usage32, but can also be adapted to run on estimated transcript counts, in order to detect DTU. Using DEXSeq on transcript counts was evaluated by Soneson et al.33, showing the benefits in FDR control from filtering lowly expressed transcripts for a transcript-level analysis. We benchmarked DEXSeq here, beginning with the DRIMSeq filtered object, as these filters are intuitive, they greatly speed up the analysis, and such filtering was shown to be beneficial in FDR control.
The two factors of (1) working on isoform counts rather than individual exons and (2) using the DRIMSeq filtering procedure dramatically increase the speed of DEXSeq, compared to running an exon-level analysis. Another advantage is that we benefit from the sophisticated bias models of Salmon, which account for drops in coverage on alternative exons that can otherwise throw off estimates of transcript abundance26. A disadvantage over the exon-level analysis is that we must know in advance all of the possible isoforms that can be generated from a gene locus, all of which are assumed to be contained in the annotation files (FASTA and GTF).
We first load the DEXSeq package and then build a DEXSeqDataSet from the data contained in the dmDStest object (the class of the DRIMSeq object changes as the results are added). The design formula of the DEXSeqDataSet here uses the language “exon” but this should be read as “transcript” for our analysis. DEXSeq will test – after accounting for total gene expression for this sample and for the proportion of this transcript relative to the others – whether there is a condition-specific difference in the transcript proportion relative to the others. The testing of “this” vs “others” in DEXSeq enables it to be much faster than its original published version, which involved fitting coefficients for each exon within a gene (here it would have been for each transcript within a gene).
library(DEXSeq) sample.data <- DRIMSeq::samples(d) count.data <- round(as.matrix(counts(d)[,-c(1:2)])) dxd <- DEXSeqDataSet(countData=count.data, sampleData=sample.data, design=~sample + exon + condition:exon, featureID=counts(d)$feature_id, groupID=counts(d)$gene_id)
The following functions run the DEXSeq analysis. While we are only working on a subset of the data, the full analysis for this dataset took less than 3 minutes on a laptop.
system.time({ dxd <- estimateSizeFactors(dxd) dxd <- estimateDispersions(dxd, quiet=TRUE) dxd <- nbinomLRT(dxd, reduced=~sample + exon) }) ## user system elapsed ## 7.084 0.064 7.184
We then extract the results table, not filtering on mean counts (as we have already conducted filtering via DRIMSeq functions). We compute a per-gene adjusted p-value, using the perGeneQValue function, which aggregates evidence from multiple tests within a gene to a single p-value for the gene and then corrects for multiple testing across genes32. Other methods for aggregative evidence from the multiple tests within genes have been discussed in a recent publication and may be substituted at this step34. Finally, we build a simple results table with the per-gene adjusted p-values.
dxr <- DEXSeqResults(dxd, independentFiltering=FALSE) qval <- perGeneQValue(dxr) dxr.g <- data.frame(gene=names(qval),qval)
For size consideration of the workflow R package, we reduce also the transcript-level results table to a simple data.frame:
columns <- c("featureID","groupID","pvalue") dxr <- as.data.frame(dxr[,columns])
Again, as we have been working with only a subset of the data, we now load the results tables that would have been generated by running DEXSeq functions on the entire dataset.
data(dex_tables)
If the stageR package has not already been loaded, we make sure to load it, and run code very similar to that used above for DRIMSeq two-stage testing, with a target alpha=0.05.
library(stageR) strp <- function(x) substr(x,1,15) pConfirmation <- matrix(dxr$pvalue,ncol=1) dimnames(pConfirmation) <- list(strp(dxr$featureID),"transcript") pScreen <- qval names(pScreen) <- strp(names(pScreen)) tx2gene <- as.data.frame(dxr[,c("featureID", "groupID")]) for (i in 1:2) tx2gene[,i] <- strp(tx2gene[,i])
The following three functions provide a table with the OFDR control described above. To repeat, the set of genes passing screening should not have more than 5% of either genes which have in fact no DTU or genes which contain a transcript with an adjusted p-value less than 5% which do not participate in DTU.
stageRObj <- stageRTx(pScreen=pScreen, pConfirmation=pConfirmation, pScreenAdjusted=TRUE, tx2gene=tx2gene) stageRObj <- stageWiseAdjustment(stageRObj, method="dtu", alpha=0.05) suppressWarnings({ dex.padj <- getAdjustedPValues(stageRObj, order=FALSE, onlySignificantGenes=TRUE) }) head(dex.padj)
## geneID txID gene transcript
## 1 ENSG00000001167 ENST00000341376 0.0000877079 0
## 2 ENSG00000001167 ENST00000353205 0.0000877079 0
## 3 ENSG00000001461 ENST00000003912 0.0051524663 0
## 4 ENSG00000001461 ENST00000339255 0.0051524663 0
## 5 ENSG00000001630 ENST00000003100 0.0234729668 0
## 6 ENSG00000001630 ENST00000450723 0.0234729668 0SUPPA2 is a command-line software package written in Python that also takes as input Salmon quantification, and so, for completeness, we also show example commands and evaluate its performance on the simulated data35. SUPPA2 offers a number of distinct features, including the ability to translate from Salmon transcript-level quantifications to individual splicing events, which are cataloged using a specific vocabulary described in the SUPPA2 software usage guide. SUPPA2 additionally offers differential analysis on the splicing events, which may be more valuable to investigators than per-transcript results, depending on the research goals (similar to the exon-level primary use case of DEXSeq).
Here, as our DTU simulation involved switching between expressed transcripts without assessing whether they were separated by one or more splice events, and as the other two Bioconductor methods for detecting DTU involve transcript-level analysis, we ran SUPPA2 in its differential transcript usage mode. We chose to filter on transcripts with TPM larger than 1; TPM filtering is a command-line option available during the diffSplice step of SUPPA2 and this improves the running time. We did not use gene-correction, as we wanted to apply the aggregation and correction method perGeneQValue from DEXSeq to obtain an FDR bounded set of genes and transcripts as output. We did not perform the stage-wise analysis of SUPPA2 output, although this could be done by small modifications to the above code for either DRIMSeq or DEXSeq.
We used the following R code to prepare two files containing TPM estimates for each of the two groups, using the tximport object defined above:
x <- txi$abundance x[x < 0.01] <- 0 # eliminate very small TPMs n <- 6 # sample size per group write.table(x[,1:n], file=paste0("suppa/group1.tpm"), quote=FALSE, sep="\t") write.table(x[,n + 1:n], file=paste0("suppa/group2.tpm"), quote=FALSE, sep="\t")
The SUPPA2 example code can be found at the software homepage, but we include here the code used on the 6 vs 6 analysis. The first line generates a set of isoforms from the GTF file. The second and third line generate PSI (percent spliced in) estimates for each transcript from files containing the TPMs for each group. The final line performs the differential analysis.
python suppa.py generateEvents -f ioi -i gencode.v28.annotation.gtf \
-o suppa/isoforms
python suppa.py psiPerIsoform -g gencode.v28.annotation.gtf \
-e suppa/group1.tpm -o suppa/group1
python suppa.py psiPerIsoform -g gencode.v28.annotation.gtf \
-e suppa/group2.tpm -o suppa/group2
python suppa.py diffSplice -m empirical -th 1 -i suppa/isoforms.ioi \
-p suppa/group1_isoform.psi suppa/group2_isoform.psi \
-e suppa/group1.tpm suppa/group2.tpm -o suppa/diff_empiricalWe imported the analysis results into R:
suppa <- read.delim("suppa/diff_empirical.dpsi") names(suppa) <- c("txp.gene","dpsi","pval") suppa$gene <- sub(";.*", "", suppa$txp.gene) suppa$txp <- sub(".*;", "", suppa$txp.gene) suppa <- suppa[!is.nan(suppa$dpsi),]
The following line was used to compute transcript-level adjusted p-values. We noticed that SUPPA2 had a large gain in sensitivity, while still controlling its FDR, if the set of transcripts examined were limited to those that passed the DRIMSeq filtering steps above. Therefore, before running any multiple test correction steps, we filtered to this subset of transcripts. We assessed whether the TPM > 1 filtering step made a difference in the sensitivity and false discovery rate for SUPPA2 when combined with the DRIMSeq filtering; it did not.
suppa <- suppa[match(res.txp$feature_id, suppa$txp),] suppa$padj <- p.adjust(suppa$pval, method="BH")
We generated per-gene adjusted p-values, using perGeneQValue from DEXSeq:
library(DEXSeq) suppa.dxr <- as(DataFrame(groupID=suppa$gene, pvalue=suppa$pval, padj=rep(1, nrow(suppa))), "DEXSeqResults") qval <- perGeneQValue(suppa.dxr) suppa.g <- data.frame(gene=names(qval), qval=qval)
We begin the evaluation by noting that all of the methods correctly avoided calling many of the DGE events as DTU events. The object dge.genes contains the names of all the genes in which all the isoforms were differentially expressed by an equal amount (so not DTU). SUPPA2 output is not included in the workflow, but it only reported one of the DGE genes as DTU out of 851 with an adjusted p-value less than 0.05.
The number of DGE genes called in DTU analysis with DRIMSeq:
res$dge <- res$gene_id %in% dge.genes with(res, table(sig=adj_pvalue < .05, dge))
## dge
## sig FALSE TRUE
## FALSE 5375 754
## TRUE 1590 17The number of DGE genes called in DTU analysis with DEXSeq:
dxr.g$dge <- dxr.g$gene %in% dge.genes with(dxr.g, table(sig=qval < .05, dge))
## dge
## sig FALSE TRUE
## FALSE 5538 769
## TRUE 1455 2The iCOBRA package36 was used to construct plots to assess the true positive rate over the false discovery rate at three nominal FDR thresholds: 1%, 5%, and 10%. The code for evaluating all methods and constructing the iCOBRA plots is included in the simulation repository21. Above, we showed an analysis for a comparison of 6 vs 6 samples. As we were interested in the performance at various sample sizes, we performed the entire analysis for DRIMSeq, DEXSeq, and SUPPA2 at per-group sample sizes of 3, 6, 9, and 12.
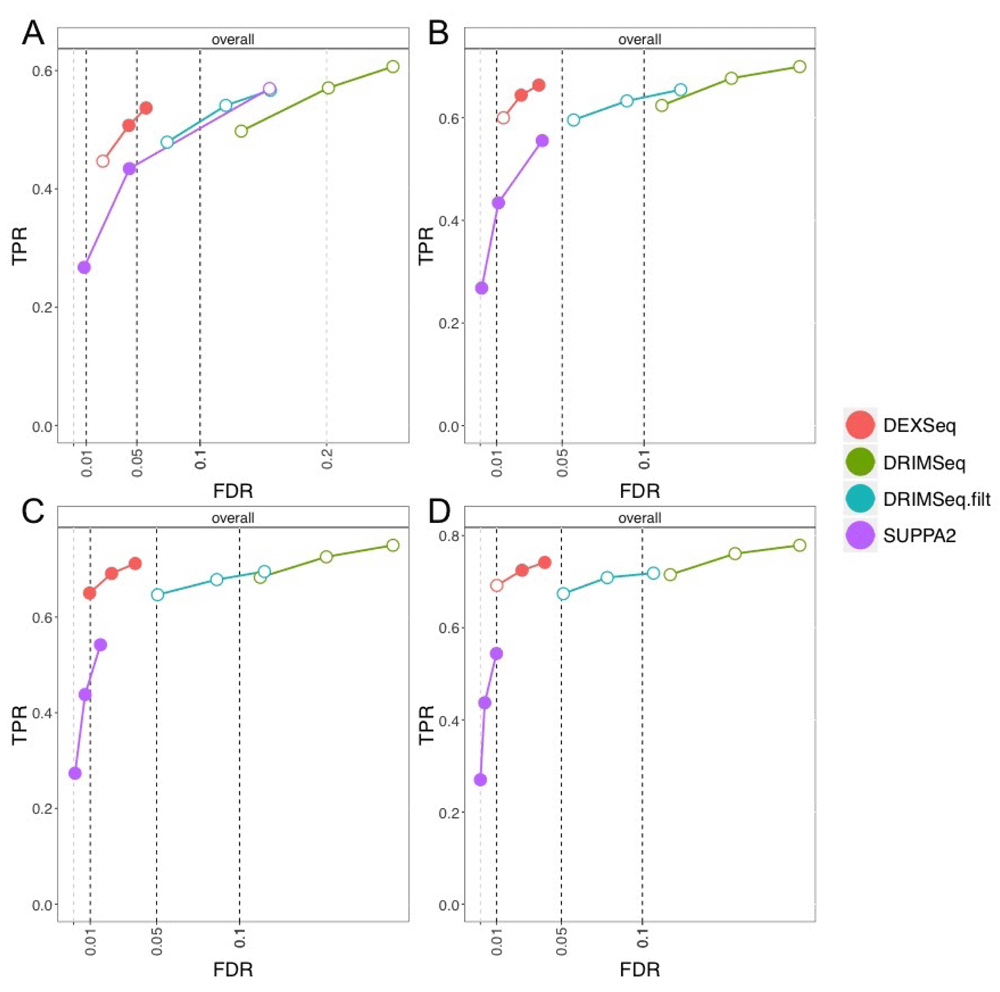
At the gene level, in terms of controlling the nominal FDR, SUPPA2 always controlled its FDR, even for the smallest sample size, DEXSeq controlled except for the 1% threshold in the smallest sample size case, and DRIMSeq exceeded its FDR but approached the target for larger sample sizes (Figure 3). Exceeding the nominal FDR level by a small amount should be considered with a method’s relative sensitivity in mind as well, compared to other methods. For example, for the 6 vs 6 comparison, DRIMSeq had observed FDR of 12% at nominal 10%, meaning that for every 100 genes reported as containing DTU, the method reported 2 extra genes more than its target. DRIMSeq and DEXSeq were the most sensitive methods in recovering gene-level DTU in this simulation.
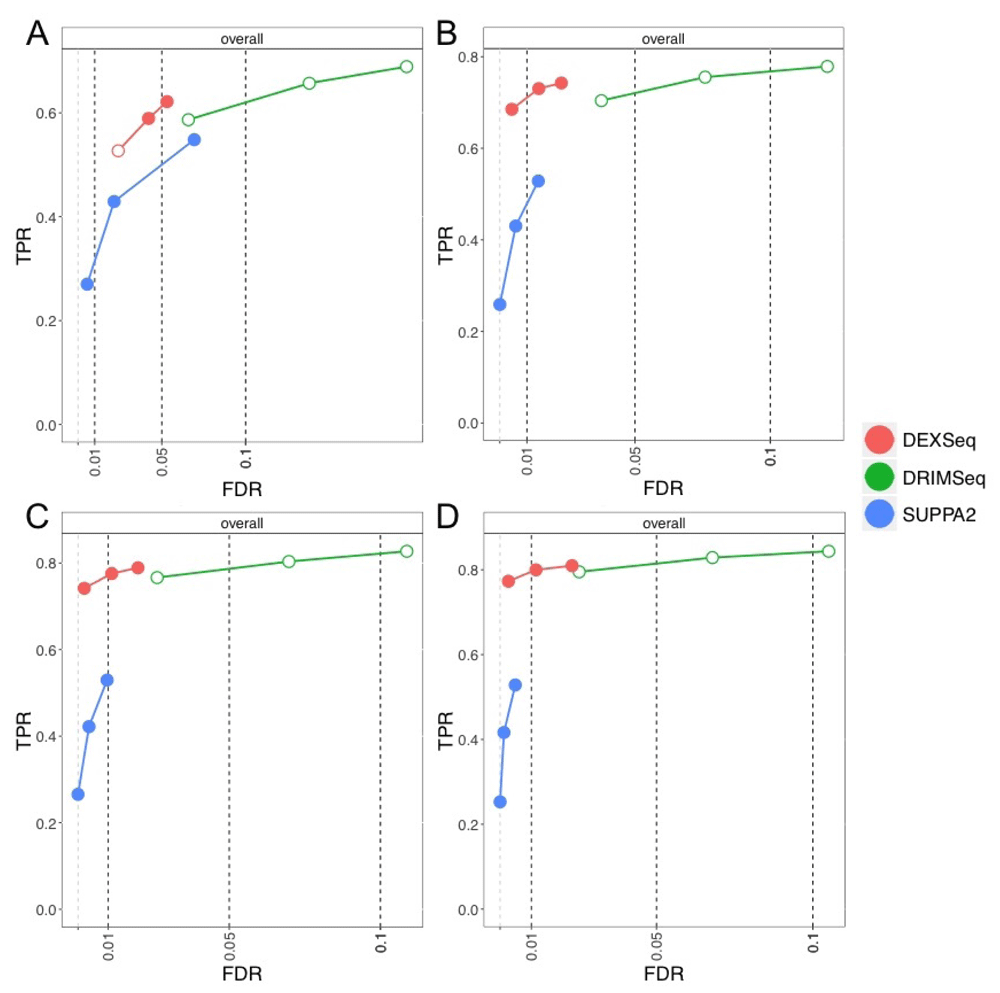
True positive rate (y-axis) over false discovery rate (FDR) (x-axis) for DEXSeq, DRIMSeq, and SUPPA2. The four panels shown are for per-group sample sizes: (A) 3, (B) 6, (C) 9, and (D) 12. Circles indicate thresholds of 1%, 5%, and 10% nominal FDR, which are filled if the observed value is less than the target (dashed vertical lines).
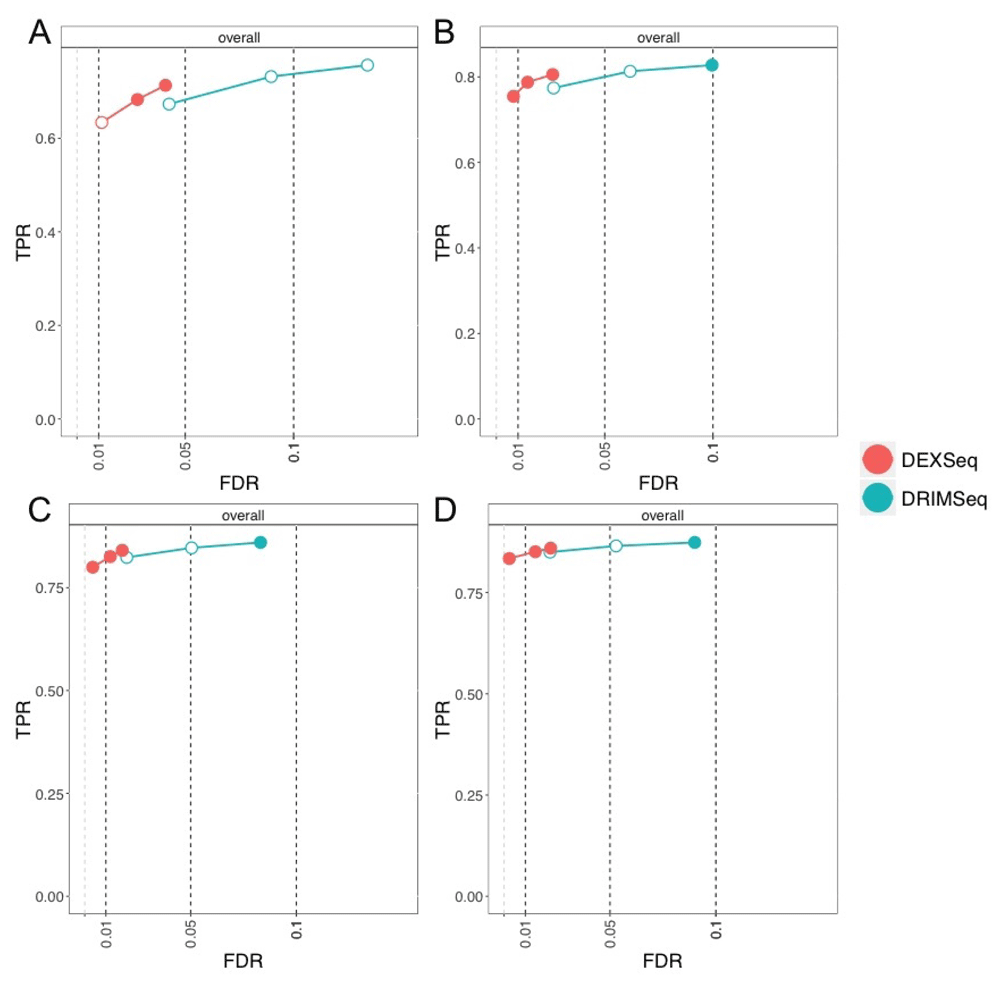
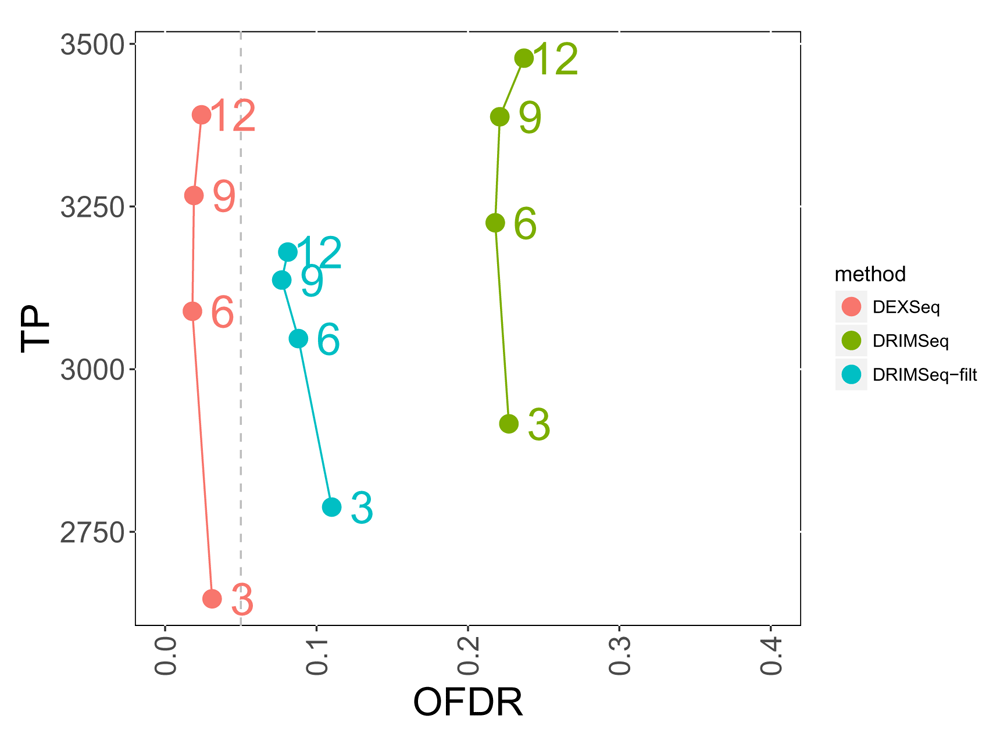
We assessed the overall false discovery rate (OFDR) procedure implemented with stageR using gene- and transcript-level p-values from DRIMSeq and DEXSeq. For DRIMSeq, we assessed whether raising the p-values for transcripts with small proportion SD helped to recover OFDR control. DEXSeq input to stageR tended to stay within the 5% OFDR target, and the observed OFDR for DRIMSeq with proportion SD filtering lowered to around 15% at per-group sample size of 6 and higher (Figure 4). Without the filtering, the observed OFDR for DRIMSeq was otherwise around 25%.
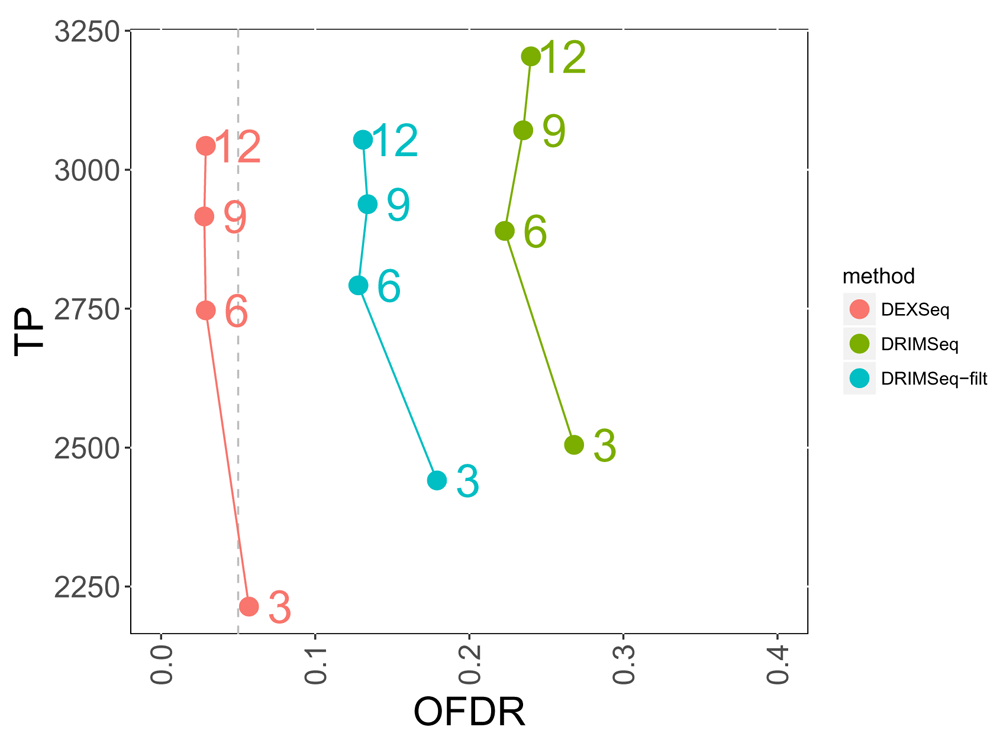
Each method is drawn as a line, and the numbers to the right of the points indicate the per-group sample size. Adjusted p-values for a nominal 5% OFDR (dashed vertical line) were generated for DEXSeq and DRIMSeq (with and without post-hoc filtering) from gene- and transcript-level p-values using the stageR framework for stage-wise testing.
Finally, we assessed the transcript-level adjusted p-values for DTU directly from DRIMSeq, DEXSeq, and SUPPA2. This analysis did not use stageR for stage-wise testing, and so we compute the standard FDR, where the unit of false discovery is the transcript, in contrast to the OFDR where the unit of false discovery is the gene. In general, we recommend using the stageR results, as it allows error control on a natural procedure of looking across genes, then within genes for which transcripts participate in DTU. SUPPA2 again tended to control its FDR, as did DEXSeq (Figure 5). DRIMSeq with proportion SD filtering approached the target FDR as sample size increased for the 5% and 10% targets, while without filtering, the observed FDR was always higher than the target.
True positive rate (y-axis) over false discovery rate (x-axis) for DEXSeq, DRIMSeq (with and without post-hoc filtering), and SUPPA2. The four panels shown are for per-group sample sizes: (A) 3, (B) 6, (C) 9, and (D) 12. Circles indicate thresholds of 1%, 5%, and 10% nominal FDR.
In Table 1 we include the timing for each method at various sample sizes. Timing includes only the diffSplice step of SUPPA2 (the other steps take less than a minute). For DRIMSeq and DEXSeq, we include the timing of the estimation steps (importing counts with tximport and filtering takes only a few seconds).
In order to further investigate performance differences between DRIMSeq and DEXSeq, we generated an additional simulation in which genes were assigned Negative Binomial dispersion parameters by matching the gene-level count to the joint distribution of mean and dispersions on the GEUVADIS dataset. Then transcript-level counts were generated with all transcripts of a gene being assigned the same Negative Binomial dispersion parameter. This contrasts with the main simulation, in which each transcript was assigned its own dispersion parameter, resulting in heterogeneity of dispersion within a gene. As we do not know the degree to which transcripts of a gene would have correlated biological variability in an experimental dataset, we also include the results for the count-based methods that estimate precision/dispersion, DRIMSeq and DEXSeq on this additional simulation.
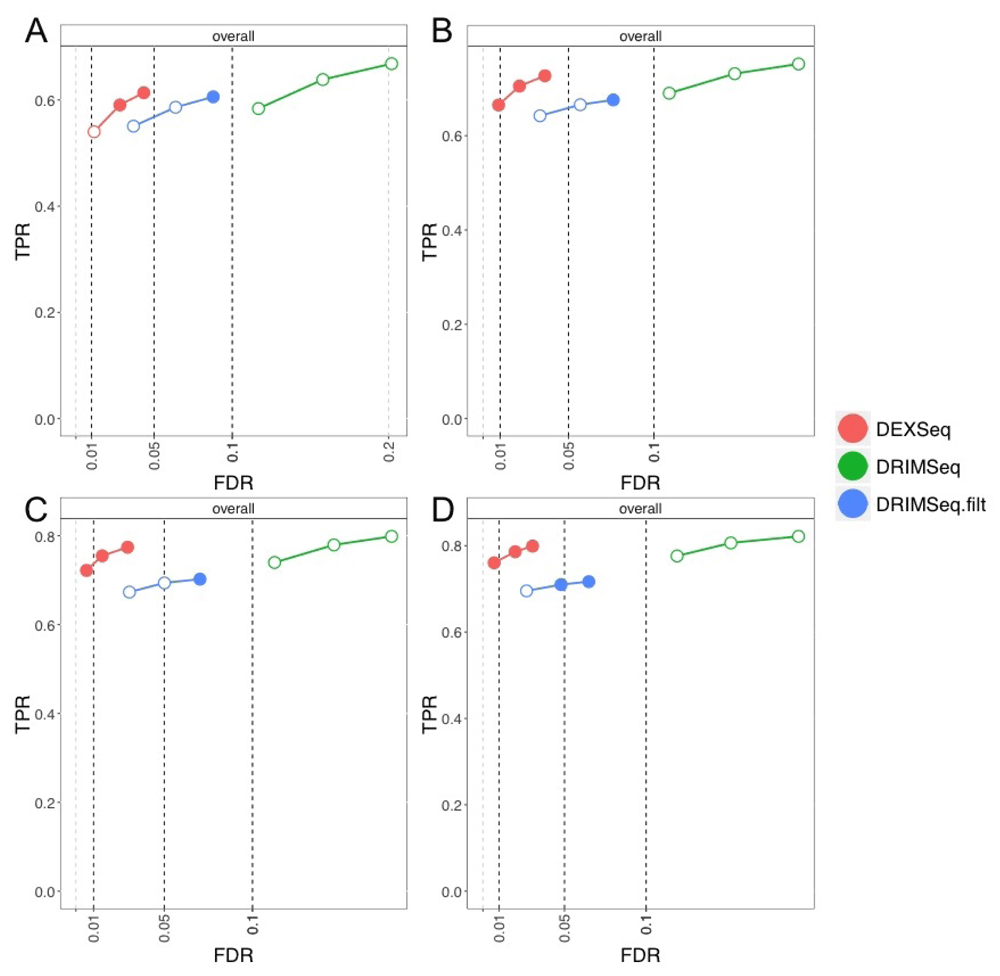
DRIMSeq, which estimates a single precision parameter per gene, performed slightly better on this simulation at the gene level (Figure 6), although we note that DRIMSeq nearly controlled FDR at the gene level already in the main simulation. DEXSeq models different dispersion parameters for every transcript, and its performance changes less across the two simulations. More improvement was seen for DRIMSeq with proportion SD filtering, in the OFDR analysis (Figure 7) and in the transcript-level analysis without screening (Figure 8). Again, we caveat our comparative evaluation of DRIMSeq and DEXSeq by noting that we do not know whether various real RNA-seq experiments will more closely reflect within-gene heterogeneous dispersion or fixed dispersion, or something in between.
The four panels shown are for per-group sample sizes: (A) 3, (B) 6, (C) 9, and (D) 12. Circles indicate thresholds of 1%, 5%, and 10% nominal FDR.
The four panels shown are for per-group sample sizes: (A) 3, (B) 6, (C) 9, and (D) 12. Circles indicate thresholds of 1%, 5%, and 10% nominal FDR.
In the final section of the workflow containing live code examples, we demonstrate how differential transcript usage, summarized to the gene-level, can be visualized with respect to differential gene expression analysis results. We use tximport and summarize counts to the gene level and compute an average transcript length offset for count-based methods12. We will then show code for using DESeq2 and edgeR to assess differential gene expression. Because we have simulated the genes according to three different categories, we can color the final plot by the true simulated state of the genes. We note that we will pair DEXSeq with DESeq2 results in the following plot, and DRIMSeq with edgeR results. However, this pairing is arbitrary, and any DTU method can reasonably be paired with any DGE method.
The following line of code is unevaluated, but was used to generate an object txi.g which contains the gene-level counts, abundances and average transcript lengths.
txi.g <- tximport(files, type="salmon", tx2gene=txdf[,2:1])
For the workflow, we load the txi.g object which is saved in a file salmon_gene_txi.rda. We then load the DESeq2 package and build a DESeqDataSet from txi.g, providing also the sample information and a design formula.
data(salmon_gene_txi) library(DESeq2) dds <- DESeqDataSetFromTximport(txi.g, samps, ~condition)
## using counts and average transcript lengths from tximport
The following two lines of code run the DESeq2 analysis16.
dds <- DESeq(dds) dres <- DESeq2::results(dds)
We can confirm that most of the DTU genes are correctly not included in the significant DGE results (although some are).
length(dtu.genes)
## [1] 1501
table(rownames(dres)[which(dres$padj < .05)] %in% dtu.genes)
##
## FALSE TRUE
## 2587 102Because we happen to know the true status of each of the genes, we can make a scatterplot of the results, coloring the genes by their status (whether DGE, DTE, or DTU by construction).
all(dxr.g$gene %in% rownames(dres))
## [1] TRUEdres <- dres[dxr.g$gene,] # we can only color because we simulated... col <- rep(8, nrow(dres)) col[rownames(dres) %in% dge.genes] <- 1 col[rownames(dres) %in% dte.genes] <- 2 col[rownames(dres) %in% dtu.genes] <- 3
Figure 9 displays the evidence for differential transcript usage over that for differential gene expression. We can see that the DTU genes cluster on the y-axis (mostly not captured in the DGE analysis), and the DGE genes cluster on the x-axis (mostly not captured in the DTU analysis). The DTE genes fall in the middle, as all of them represent DGE, and some of them additionally represent DTU (if the gene had other expressed transcripts). Because DEXSeq outputs an adjusted p-value of 0 for some of the genes, we set these instead to a jittered value around 10−20, so that their number and location on the x-axis could be visualized. These jittered values should only be used for visualization.
bigpar() # here cap the smallest DESeq2 adj p-value cap.padj <- pmin(-1og10(dres$padj), 100) # this vector only used for plotting jitter.padj <- -1og10(dxr.g$qval + 1e-20) jp.idx <- jitter.padj == 20 jitter.padj[jp.idx] <- rnorm(sum(jp.idx),20,.25) plot(cap.padj, jitter.padj, col=col, xlab="Gene expression", ylab="Transcript usage") legend("topright", c("DGE","DTE","DTU","null"), col=c(1:3,8), pch=20, bty="n")
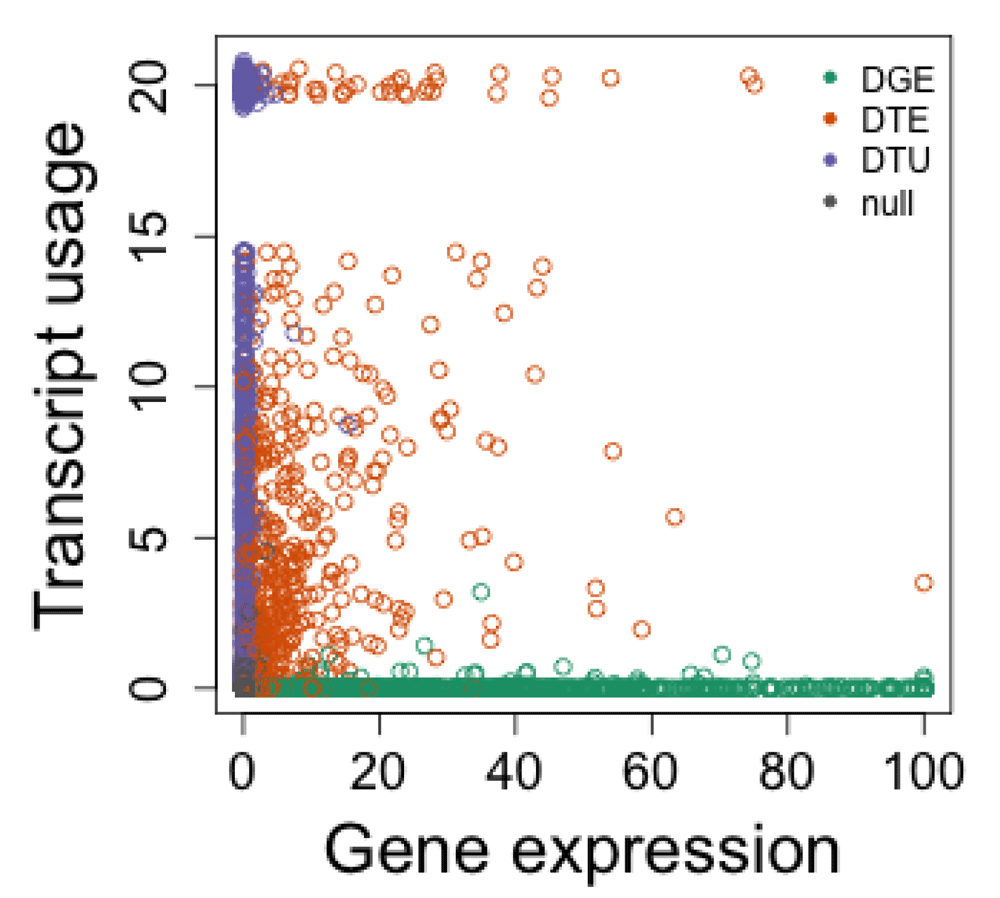
Each point represents a gene, and plotted are -log10 adjusted p-values for DEXSeq’s test of differential transcript usage (y-axis) and DESeq2’s test of differential gene expression (x-axis). Because we simulated the data we can color the genes according to their true category.
We can repeat the same analysis using edgeR as the inference engine3. The following code incorporates the average transcript length matrix as an offset for an edgeR analysis.
library(edgeR) cts.g <- txi.g$counts normMat <- txi.g$length normMat <- normMat / exp(rowMeans(log(normMat))) o <- log(calcNormFactors(cts.g/normMat)) + log(colSums(cts.g/normMat)) y <- DGEList(cts.g) y <- scaleOffset(y, t(t(log(normMat)) + o)) keep <- filterByExpr(y) y <- y[keep,]
The basic edgeR model fitting and results extraction can be accomplished with the following lines:
y <- estimateDisp(y, design_full) fit <- glmFit(y, design_full) lrt <- glmLRT(fit) tt <- topTags(lrt, n=nrow(y), sort="none")[[1]]
We confirm that most of the DTU genes are correctly not reported as DGE:
table(rownames(tt)[which(tt$FDR < .05)] %in% dtu.genes)
##
## FALSE TRUE
## 2280 31Again, we can color the genes by their true status in the simulation:
common <- intersect(res$gene_id, rownames(tt)) tt <- tt[common,] res.sub <- res[match(common, res$gene_id),] # we can only color because we simulated... col <- rep(8, nrow(tt)) col[rownames(tt) %in% dge.genes] <- 1 col[rownames(tt) %in% dte.genes] <- 2 col[rownames(tt) %in% dtu.genes] <- 3
Figure 10 displays the evidence for differential transcript usage over that for differential gene expression, now using DRIMSeq and edgeR. One obvious contrast with Figure 9 is that DRIMSeq outputs lower non-zero adjusted p-values than DEXSeq does, where DEXSeq instead outputs 0 for many genes. The plots look more similar when zooming in on the DRIMSeq y-axis, as can be seen in Figure 11.
bigpar() plot(-log10(tt$FDR), -log10(res.sub$adj_pvalue), col=col, xlab="Gene expression", ylab="Transcript usage") legend("topright", c("DGE","DTE","DTU","null"), col=c(1:3,8), pch=20, bty="n")
bigpar() plot(-log10(tt$FDR), -log10(res.sub$adj_pvalue), col=col, xlab="Gene expression", ylab="Transcript usage", ylim=c(0,20)) legend("topright", c("DGE","DTE","DTU","null"), col=c(1:3,8), pch=20, bty="n")
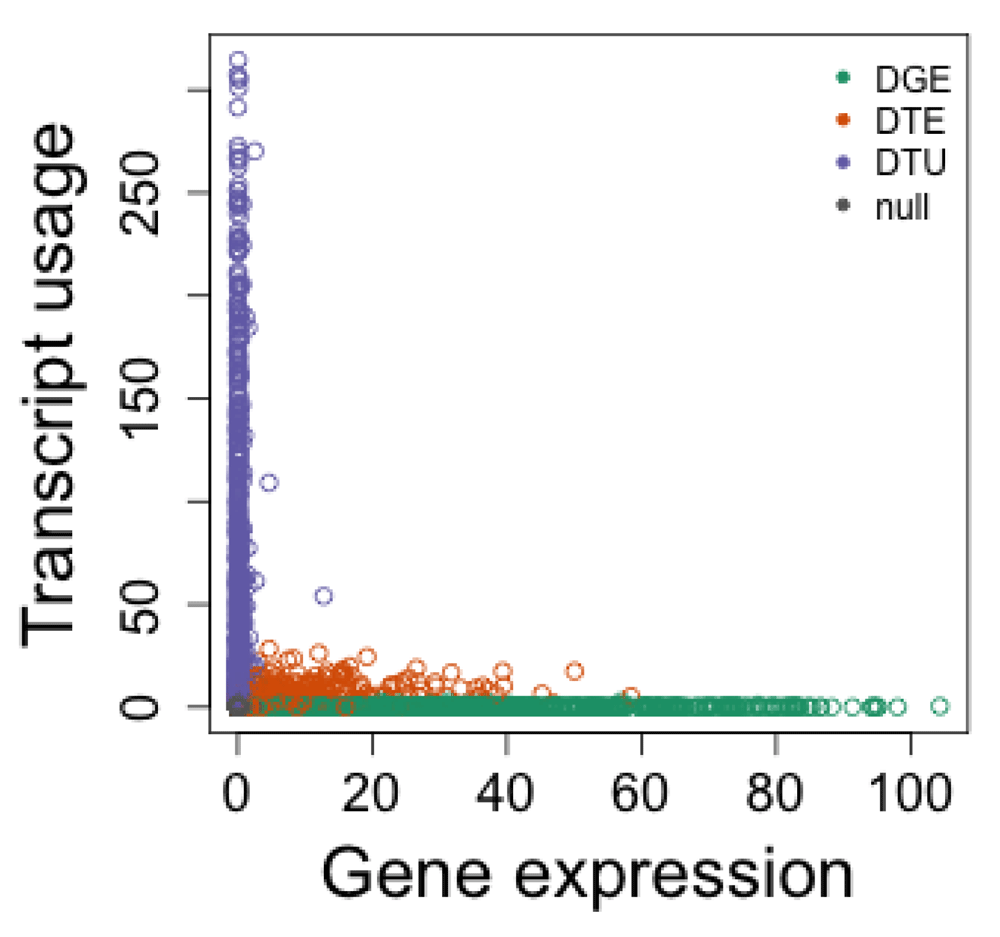
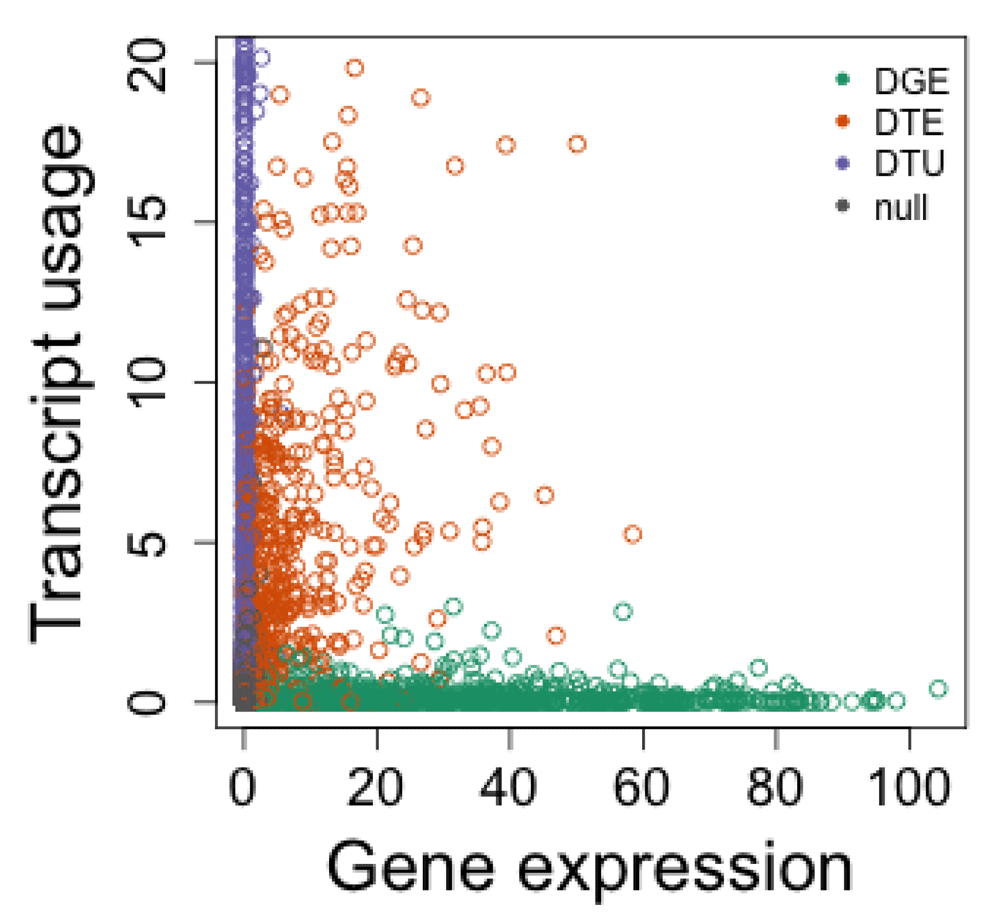
We additionally assessed Bioconductor and other R packages for differential gene expression, to determine true positive rate and control of false discovery rate on the simulated dataset. In this analysis, the simulated “DTE” genes (where a single transcript was chosen to be differentially expressed) should count for differential gene expression, while the simulated “DTU” genes should not, as the total expression of the gene remains constant.
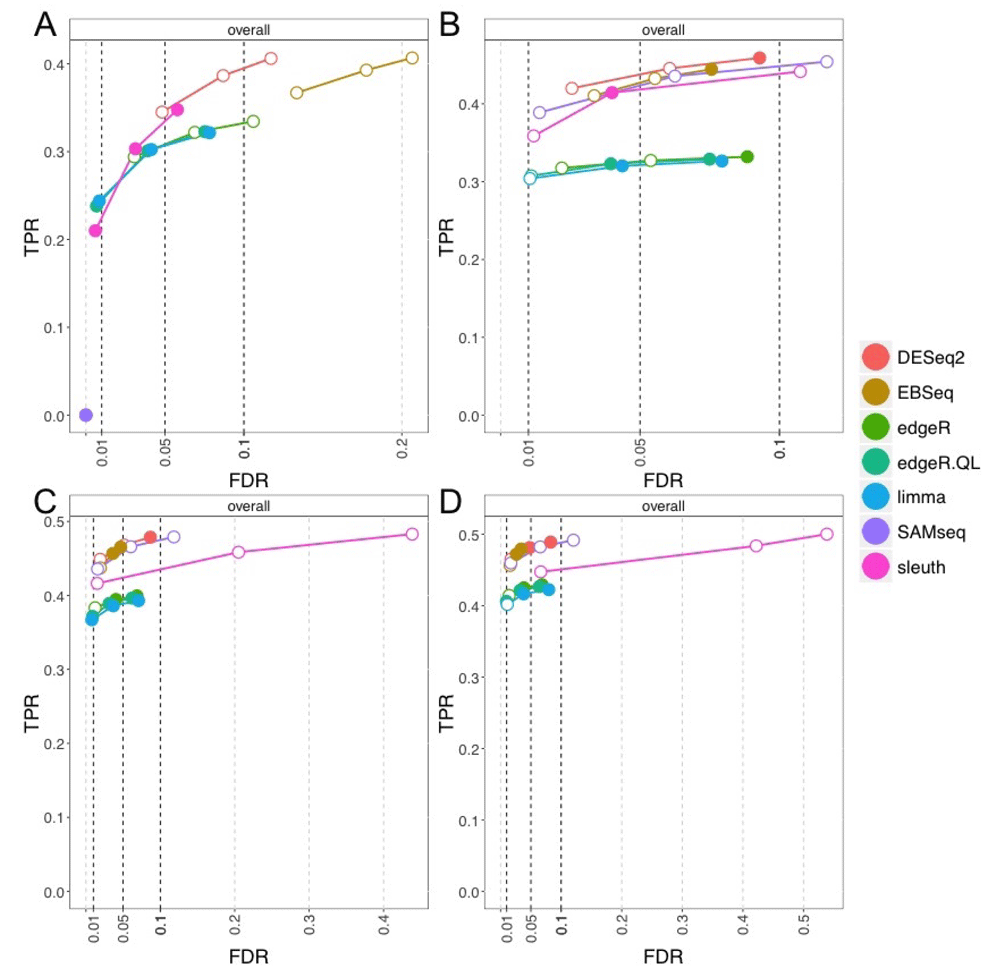
We compared DESeq216, EBSeq37, edgeR3, edgeR-QL (using the quasi-likelihood functions)38, limma with voom transformation6, SAMseq39, and sleuth40. We used tximport to summarize Salmon abundances to the gene level, and provided all methods other than DESeq2 and sleuth with the lengthScaledTPM count matrix. sleuth takes as input the quantification from kallisto15, which was run with 30 bootstrap samples and bias correction. For gene-level analysis in sleuth, the argument aggregation_column="gene_id" was used. As DESeq2 has specially designed import functions for taking in estimated gene counts and an offset from tximport, we used this approach to provide Salmon summarized gene-level counts and an offset. edgeR and edgeR-QL had the same performance using the counts and offset approach or the lengthScaledTPM approach, so we used the latter for code simplicity. The exact code used to run the different methods can be found at the simulation code repository21. Timings for the different gene-level methods are presented in Table 2.
Timing includes data import and summarization to gene-level quantities using one core.
| Method | n=3 | n=6 | n=9 | n=12 |
|---|---|---|---|---|
| DESeq2 | <1 | <1 | <1 | <1 |
| EBSeq | 1 | 2 | 2 | 3 |
| edgeR | <1 | <1 | <1 | <1 |
| edgeR-QL | <1 | <1 | <1 | <1 |
| limma | <1 | <1 | <1 | <1 |
| SAMseq | <1 | <1 | <1 | <1 |
| sleuth | 2 | 4 | 5 | 7 |
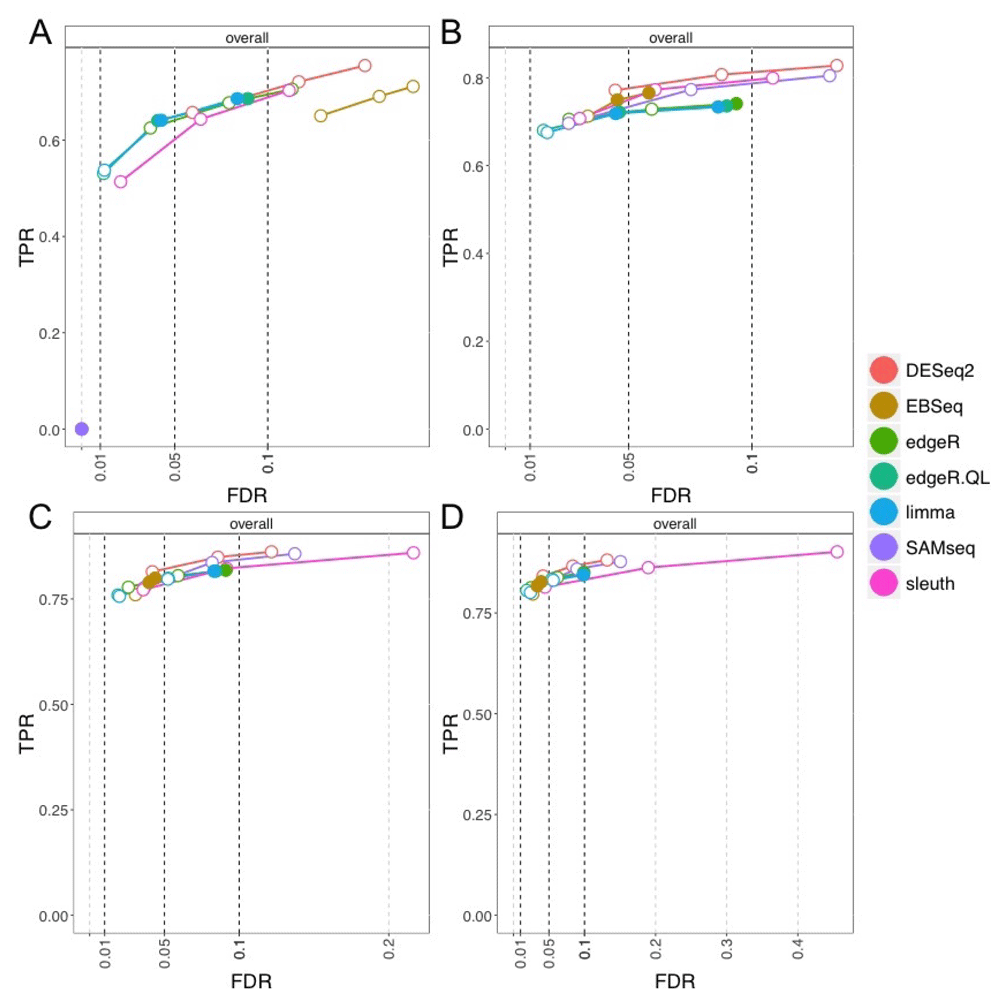
iCOBRA plots with true positive rate over false discovery rate for gene-level analysis across four different per-group sample sizes are presented in Figure 12. For the smallest per-group sample size of 3, all methods except DESeq2 and EBSeq tended to control the FDR, while those two method had, for example, 15% FDR at the nominal 10% rate. SAMseq, with so few samples, did not have any sensitivity to detect DGE. At the per-group sample size of 6, all methods except DESeq2 and SAMseq tended to control the FDR. At this sample size, EBSeq controlled its FDR. For the largest per-group sample sizes, 9 and 12, the performance of many methods remained similar as previously, except sleuth did not control the nominal 5% or 10% FDR. We performed additional experiments to see if the performance of sleuth at higher sample sizes was related to the realistic GC bias parameters used in the simulation, but simulating fragments uniformly from the transcripts revealed the same performance at per-group sample sizes 9 and 12 (Supplementary Figure 2). Reducing the number of DGE, DTE and DTU genes from 10% to 5% each, however, did recover control of the FDR at the nominal 5% and 10% FDR for sleuth (Supplementary Figure 3).
Finally, we assessed the Bioconductor and R packages for differential transcript expression analysis. While we believe the separation of differential transcript usage and differential gene expression described in the earlier sections of the workflow represents an easily interpretable approach, some investigators may prefer to assess differential expression on a per-transcript basis. For this assessment, all of the simulated non-null transcripts count as DTE, whether from the simulated DGE-, DTE-, or DTU-by-construction genes. For most of the methods, we simply provided the transcript-level data to the same functions as for the DGE analysis. EBSeq was provided with the number of isoforms per gene. The timing of the methods is presented in Table 3.
Timing includes data import.
| Method | n=3 | n=6 | n=9 | n=12 |
|---|---|---|---|---|
| DESeq2 | <1 | <1 | <1 | 1 |
| EBSeq | 5 | 11 | 18 | 22 |
| edgeR | <1 | <1 | <1 | <1 |
| edgeR-QL | <1 | <1 | <1 | <1 |
| limma | <1 | <1 | <1 | <1 |
| SAMseq | <1 | <1 | <1 | 1 |
| sleuth | 2 | 2 | 2 | 2 |
iCOBRA plots with the true positive rate over false discovery rate for the transcript-level analysis are shown in Figure 13. The performance at per-group sample size of 3 was similar to the gene-level analysis, except DESeq2 came closer to controlling the FDR and EBSeq performed slightly worse than before, while the rest of the methods tended to control their FDR. At per-group sample size of 6, all of the evaluated methods tended to control the FDR, though DESeq2, EBSeq, SAMseq, and sleuth tended to have higher sensitivity than edgeR, edgeR-QL and limma. The same issue of FDR control for sleuth was seen in the transcript-level analysis as in the gene-level analysis, for per-group sample size 9 and 12.
Here we presented a workflow for analyzing RNA-seq experiments for differential transcript usage across groups of samples. The Bioconductor packages used, DRIMSeq, DEXSeq, and stageR, are simple to use and fast when run on transcript-level data. We show how these can be used downstream of transcript abundance quantification with Salmon. We evaluated these methods on a simulated dataset and showed how the transcript usage results complement a gene-level analysis, which can also be run on output from Salmon, using the tximport package to aggregate quantification to the gene level. We used the simulated dataset to evaluate Bioconductor and other R packages for differential gene expression, and differential transcript expression. We recommend the use of stageR for its formal statistical procedure involving a screening and confirmation stage, as this fits closely to what we expect a typical analysis to entail. stageR then provides error control for an overall false discovery rate, assuming that the underlying tests are well calibrated.
One potential limitation of this workflow is that, in contrast to other methods such as the standard DEXSeq analysis, SUPPA2, or LeafCutter41, here we considered and detected expression switching between annotated transcripts. Other methods such as DEXSeq (exon-based), SUPPA2, or LeafCutter may benefit in terms of power and interpretability from performing statistical analysis directly on exon usage or splice events. Methods such as DEXSeq (exon-based) and LeafCutter benefit in the ability to detect un-annotated events. The workflow presented here would require further processing to attribute transcript usage changes to specific splice events, and is limited to considering the estimated abundance of annotated transcripts.
The following provides the session information used when compiling this document.
devtools::session_info()
## Session info -------------------------------------------------------------
## setting value
## version R version 3.5.0 (2018-04-23)
## system x86_64, darwin15.6.0
## ui X11
## language (EN)
## collate en_US.UTF-8
## tz America/New_York
## date 2018-06-17
## Packages -----------------------------------------------------------------
## package * version date source
## acepack 1.4.1 2016-10-29 CRAN (R 3.5.0)
## annotate 1.58.0 2018-05-01 Bioconductor
## AnnotationDbi * 1.42.1 2018-05-08 Bioconductor
## assertthat 0.2.0 2017-04-11 CRAN (R 3.5.0)
## backports 1.1.2 2017-12-13 cran (@1.1.2)
## base * 3.5.0 2018-04-24 local
## base64enc 0.1-3 2015-07-28 CRAN (R 3.5.0)
## Biobase * 2.40.0 2018-05-01 Bioconductor
## BiocGenerics * 0.26.0 2018-05-01 Bioconductor
## BiocInstaller * 1.30.0 2018-05-04 Bioconductor
## BiocParallel * 1.14.1 2018-05-06 Bioconductor
## BiocStyle 2.8.0 2018-05-01 Bioconductor
## BiocWorkflowTools 1.6.1 2018-05-24 Bioconductor
## biomaRt 2.36.0 2018-05-01 Bioconductor
## Biostrings 2.48.0 2018-05-01 Bioconductor
## bit 1.1-12 2014-04-09 CRAN (R 3.5.0)
## bit64 0.9-7 2017-05-08 CRAN (R 3.5.0)
## bitops 1.0-6 2013-08-17 CRAN (R 3.5.0)
## blob 1.1.1 2018-03-25 CRAN (R 3.5.0)
## bookdown 0.7 2018-02-18 CRAN (R 3.5.0)
## checkmate 1.8.5 2017-10-24 CRAN (R 3.5.0)
## cluster 2.0.7-1 2018-04-13 CRAN (R 3.5.0)
## codetools 0.2-15 2016-10-05 CRAN (R 3.5.0)
## colorspace 1.3-2 2016-12-14 CRAN (R 3.5.0)
## compiler 3.5.0 2018-04-24 local
## data.table 1.11.2 2018-05-08 CRAN (R 3.5.0)
## datasets * 3.5.0 2018-04-24 local
## DBI 1.0.0 2018-05-02 CRAN (R 3.5.0)
## DelayedArray * 0.6.0 2018-05-01 Bioconductor
## DESeq2 * 1.20.0 2018-05-01 Bioconductor
## devtools * 1.13.5 2018-02-18 CRAN (R 3.5.0)
## DEXSeq * 1.26.0 2018-05-01 Bioconductor
## digest 0.6.15 2018-01-28 cran (@0.6.15)
## DRIMSeq * 1.8.0 2018-05-01 Bioconductor
## edgeR * 3.22.2 2018-05-24 cran (@3.22.2)
## evaluate 0.10.1 2017-06-24 CRAN (R 3.5.0)
## foreign 0.8-70 2017-11-28 CRAN (R 3.5.0)
## Formula 1.2-3 2018-05-03 CRAN (R 3.5.0)
## genefilter 1.62.0 2018-05-01 Bioconductor
## geneplotter 1.58.0 2018-05-01 Bioconductor
## GenomeInfoDb * 1.16.0 2018-05-01 Bioconductor
## GenomeInfoDbData 1.1.0 2018-01-10 Bioconductor
## GenomicRanges * 1.32.2 2018-05-06 Bioconductor
## ggplot2 2.2.1 2016-12-30 CRAN (R 3.5.0)
## git2r 0.21.0 2018-01-04 CRAN (R 3.5.0)
## graphics * 3.5.0 2018-04-24 local
## grDevices * 3.5.0 2018-04-24 local
## grid 3.5.0 2018-04-24 local
## gridExtra 2.3 2017-09-09 CRAN (R 3.5.0)
## gtable 0.2.0 2016-02-26 CRAN (R 3.5.0)
## Hmisc 4.1-1 2018-01-03 CRAN (R 3.5.0)
## htmlTable 1.11.2 2018-01-20 CRAN (R 3.5.0)
## htmltools 0.3.6 2017-04-28 CRAN (R 3.5.0)
## htmlwidgets 1.2 2018-04-19 CRAN (R 3.5.0)
## httr 1.3.1 2017-08-20 CRAN (R 3.5.0)
## hwriter 1.3.2 2014-09-10 CRAN (R 3.5.0)
## IRanges * 2.14.9 2018-05-15 Bioconductor
## knitr * 1.20 2018-02-20 CRAN (R 3.5.0)
## labeling 0.3 2014-08-23 CRAN (R 3.5.0)
## lattice 0.20-35 2017-03-25 CRAN (R 3.5.0)
## latticeExtra 0.6-28 2016-02-09 CRAN (R 3.5.0)
## lazyeval 0.2.1 2017-10-29 CRAN (R 3.5.0)
## limma * 3.36.1 2018-05-05 Bioconductor
## locfit 1.5-9.1 2013-04-20 CRAN (R 3.5.0)
## magrittr 1.5 2014-11-22 CRAN (R 3.5.0)
## Matrix 1.2-14 2018-04-13 CRAN (R 3.5.0)
## matrixStats * 0.53.1 2018-02-11 CRAN (R 3.5.0)
## memoise 1.1.0 2017-04-21 CRAN (R 3.5.0)
## methods * 3.5.0 2018-04-24 local
## munsell 0.4.3 2016-02-13 CRAN (R 3.5.0)
## nnet 7.3-12 2016-02-02 CRAN (R 3.5.0)
## parallel * 3.5.0 2018-04-24 local
## pillar 1.2.2 2018-04-26 CRAN (R 3.5.0)
## plyr 1.8.4 2016-06-08 CRAN (R 3.5.0)
## prettyunits 1.0.2 2015-07-13 CRAN (R 3.5.0)
## progress 1.1.2 2016-12-14 CRAN (R 3.5.0)
## R6 2.2.2 2017-06-17 CRAN (R 3.5.0)
## rafalib * 1.0.0 2015-08-09 CRAN (R 3.5.0)
## RColorBrewer * 1.1-2 2014-12-07 CRAN (R 3.5.0)
## Rcpp 0.12.17 2018-05-18 cran (@0.12.17)
## RCurl 1.95-4.10 2018-01-04 CRAN (R 3.5.0)
## reshape2 1.4.3 2017-12-11 CRAN (R 3.5.0)
## rlang 0.2.1 2018-05-30 cran (@0.2.1)
## rmarkdown * 1.9 2018-03-01 CRAN (R 3.5.0)
## rnaseqDTU * 0.1.0 2018-06-18 local (mikelove/rnaseqDTU@NA)
## rpart 4.1-13 2018-02-23 CRAN (R 3.5.0)
## rprojroot 1.3-2 2018-01-03 cran (@1.3-2)
## Rsamtools 1.32.0 2018-05-01 Bioconductor
## RSQLite 2.1.1 2018-05-06 CRAN (R 3.5.0)
## rstudioapi 0.7 2017-09-07 CRAN (R 3.5.0)
## S4Vectors * 0.18.1 2018-05-02 Bioconductor
## scales 0.5.0 2017-08-24 CRAN (R 3.5.0)
## splines 3.5.0 2018-04-24 local
## stageR * 1.2.22 2018-06-14 cran (@1.2.22)
## statmod 1.4.30 2017-06-18 CRAN (R 3.5.0)
## stats * 3.5.0 2018-04-24 local
## stats4 * 3.5.0 2018-04-24 local
## stringi 1.2.2 2018-05-02 CRAN (R 3.5.0)
## stringr 1.3.1 2018-05-10 CRAN (R 3.5.0)
## SummarizedExperiment * 1.10.1 2018-05-11 Bioconductor
## survival 2.42-3 2018-04-16 CRAN (R 3.5.0)
## tibble 1.4.2 2018-01-22 CRAN (R 3.5.0)
## tinytex 0.5 2018-04-16 CRAN (R 3.5.0)
## tools 3.5.0 2018-04-24 local
## utils * 3.5.0 2018-04-24 local
## withr 2.1.2 2018-03-15 CRAN (R 3.5.0)
## xfun 0.1 2018-01-22 CRAN (R 3.5.0)
## XML 3.98-1.11 2018-04-16 CRAN (R 3.5.0)
## xtable 1.8-2 2016-02-05 CRAN (R 3.5.0)
## XVector 0.20.0 2018-05-01 Bioconductor
## yaml 2.1.19 2018-05-01 CRAN (R 3.5.0)
## zlibbioc 1.26.0 2018-05-01 BioconductorThe statistical methods were evaluated using the following software versions: DRIMSeq - 1.8.0, DEXSeq - 1.26.0, stageR - 1.2.21, tximport - 1.8.0, DESeq2 - 1.20.0, EBSeq - 1.20.0, edgeR - 3.22.2, limma - 3.36.1, samr - 2.0, sleuth - 0.29.0, SUPPA2 - 2.3. The samples were quantified with Salmon version 0.10.0 and kallisto version 0.44.0. polyester version 1.16.0 and alpine version 1.6.0 were used in generating the simulated dataset.
The simulated paired-end read FASTQ files have been uploaded in three batches of eight samples each to Zenodo -
https://doi.org/10.5281/zenodo.129137522
https://doi.org/10.5281/zenodo.129140423
https://doi.org/10.5281/zenodo.129144324
The quantification files are also available as a separate Zenodo dataset - https://doi.org/10.5281/zenodo.129152225
The scripts used to generate the simulated dataset are available at the simulation GitHub repository (https://github.com/mikelove/swimdown/tree/v1.0) and archived here - https://doi.org/10.5281/zenodo.129389921. All data is available under a CC BY 4.0 license.
1. All software used in this workflow is available as part of Bioconductor version 3.7.
2. Source code for the workflow: https://github.com/mikelove/rnaseqDTU
3. Link to archived source code as at time of publication: https://doi.org/10.5281/zenodo.129391442
4. License: Artistic-2.0
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)