Keywords
Papillomaviruses, cervical cancer, oncogenic risk, translation terminatio-reinitiation, machine learning
Papillomaviruses, cervical cancer, oncogenic risk, translation terminatio-reinitiation, machine learning
Persistent infections with carcinogenic human papillomaviruses (HPVs) are the main cause of cervical neoplasia and cancer, with over 99% of the cervical lesions containing HPV sequences1–3. More than 160 HPV types have been characterized4, of which approximately a third are predominantly detected in the cervical epithelium and belong to the Alphapapillomavirus genus5,6. The viruses of this genus are further grouped into high-risk (HR) and low-risk (LR) HPV types based on their association with cervical cancer and pre-cancerous lesions7,8. Most of the HR-HPV variants belong to the Human papillomavirus 16 (alpha-9) or Human papillomavirus 18 (alpha-7) species groups9.
Phylogenetic trees constructed from alignments of complete HPV genomes cluster all oncogenic types together, suggesting a common ancestor for the HR-HPVs. However, in separate trees built from different regions of the genome, the carcinogenic potential co-segregates with the early but not with the late genes10,11. The early HPV proteins E6 and E7 have transforming properties12–14 and are required for the malignant conversion. The involvement of these proteins in tumorigenesis is thought to stem from their interactions with the tumor suppressors p53 and pRB, respectively, as well as other proteins involved in tumorigenesis14–17. Variations in E6 and E7 proteins have been suggested to determine the oncogenic potential of HPV18,19 but the potential discriminating features of the oncogenic variants are frequently observed in LR-HPVs as well20–24. Currently, the most notable molecular feature that distinguishes HR from LR-HPV is the presence of a PDZ-domain recognition motif at the extreme C terminus of the HR-E6 oncoprotein, as opposed to LR-E625–27, enabling interactions with numerous proteins that contain the PDZ domain28–30.
Identification of signatures of the HR-HPV genotypes that differentiate them from the majority of alphapapillomaviruses that lack oncogenic potential could help elucidate the genetic basis of the carcinogenic properties of HPVs, thus contributing to a better understanding of the biological mechanisms exploited by the virus to trigger neoplasia. However, at present, genomic determinants of the HPV oncogenic risk are not well understood, and the exact nature of the genetic changes that led to the emergence of the HR-HPV oncogenicity remains unknown.
To better understand HPV carcinogenesis, we revisited the search for specific genomic determinants of HR-HPV types and identified a previously unreported insert of 30 to 60 basepairs (bp) at the 3’-end of the E6 oncoprotein coding region that is present in all HR-HPV types but not in LR-HPV. This insert introduces a new stop codon, separating the nucleotide sequence coding E6 from that coding for E7, eliminating the overlap between E6 and E7 that is characteristic of the LR-HPV types. The insert confers a PDZ binding motif at the end of E6 oncoprotein which is likely important for the oncogenic potentiation. Additionally, it locates the E6 termination codon upstream of and in close proximity to the E7 initiation codon. Furthermore, the insert contains sequences complementary to human 18S rRNA in the regions of hairpins 26 and 27 that are known interactors of 40S ribosomal subunits and viral RNAs, specifically involved in IRES binding. The folding of these regions of rRNA complementarity in E6-E7 mRNAs is typically relaxed in the predicted optimal and sub-optimal secondary structures of HR-HPV strains. We hypothesize that the insert into the E6 coding sequence identified here was the primary cause of the emergence of high oncogenic potential alpha-HPV.
The complete nucleotide sequences and the amino acid sequences of HPV E1, E2, E4, E5, E6, E7, L1 and L2 proteins were collected for all sequenced alpha-HPV strains (Extended data: Table S131). We then constructed a global multiple sequence alignment of the whole genome nucleotide sequences and the amino acid sequences alignments for each protein. Maximum likelihood phylogenetic analysis of these alpha-HPVs, based on the complete nucleotide sequence, as well as the amino acid sequences of most early proteins, identified HR-HPV as a clade, whereas phylogenies of E4, E5, L1 and L2 did not support the monophyly of the HR-HPV (Figure 1; Extended data: Figure S131), in agreement with previous findings9,10. These observations are compatible with a major role of recombination in the HPV evolution.
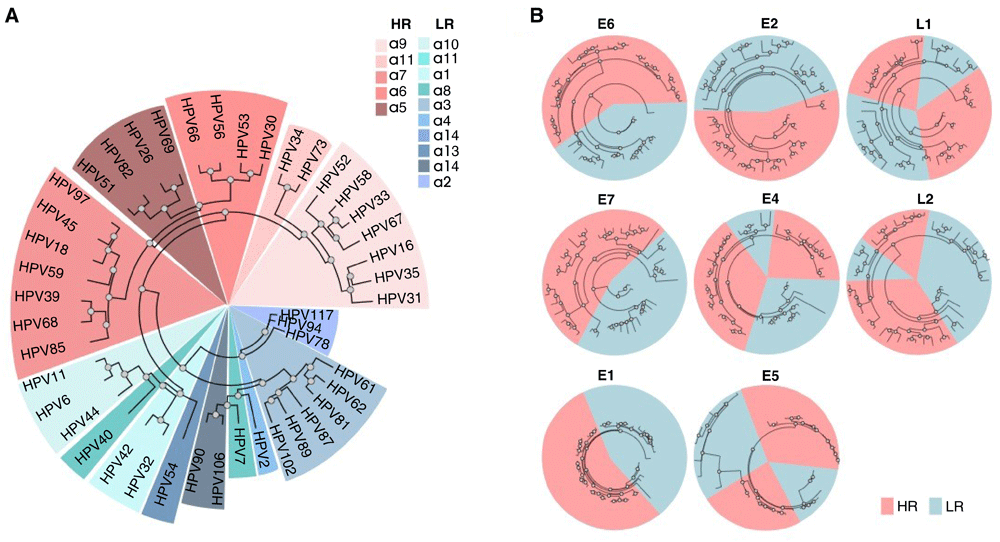
(A) Maximum likelihood tree obtained with the whole genome nucleotide sequences alignment of alpha-HPV, colored by the different alpha-class categories. (B) Maximum likelihood trees obtained with alignments of E6, E7, E1, E2, E4, E5, L1 and L2 amino acid sequences of alpha-HPV, colored by the oncogenic risk groups.
We next sought to identify genomic features that might partition alpha-HPV species, in accord with their oncogenic risk, focusing on the E6 and E7 oncogenes.
First, we searched for regions of insertions and deletions within the genome nucleotide sequences of E6 and E7 that might differentiate between the risk groups. Specifically, we identified sequences with high frequency of deletions or insertions that are located within high confidence alignment regions (See Methods for details). We then applied Support Vector Machine (SVM) with a leave-one-out cross-validation, aiming to identify regions that classify HPV strains based on their oncogenic risk. This approach resulted in the identification of genomic regions that separated HR-HPV from LR-HPV with high accuracy (over 0.75, with statistical significance; see Methods). Among these, we found one prominent insert (approximately 30-60 nucleotides) located within the E6 gene (Figure 2A). We also performed a similar search for regions separating HR-HPV from LR-HPV, using the amino acid sequence of E6 and E7 oncoproteins. For the purpose of classification, we coded the amino acids with numbers based on their frequencies and the BLOSUM6232 matrix (see Methods). As expected, the divergent region in E6 was identified from the amino acid sequences as well (Figure 2B). In contrast, we did not find any significant differences in E7 protein sequences between the high risk and low risk HPV strains (Extended data: Figure S231).
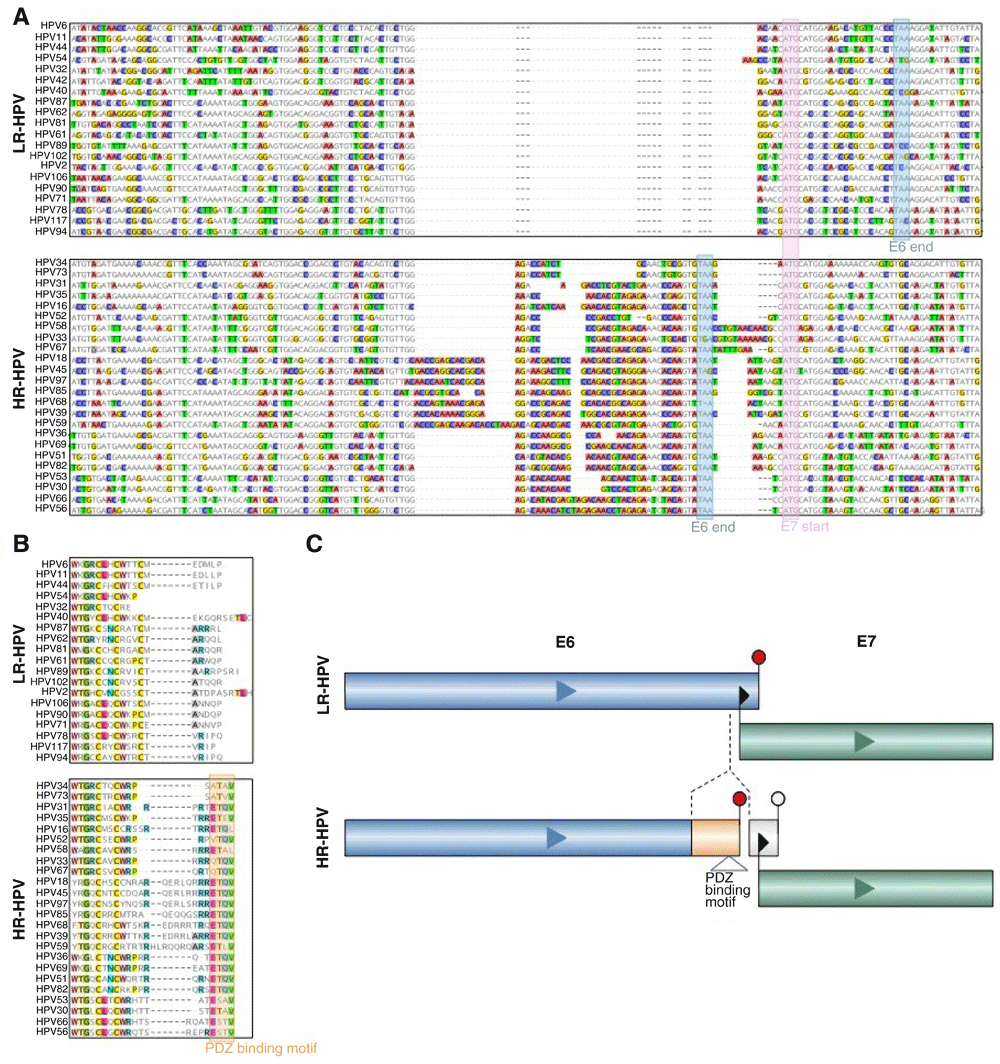
(A) Nucleotide sequence alignment of alpha-HPVs (upper panel are LR-HPV and lower panel are HR-HPV sequences). Boxed sequences: pink box, E7 start codon for most HPV types; blue boxes, E6 stop codons that are distinct between HR-HPV and LR-HPV. (B) Amino acid sequence alignment of alpha-HPVs (upper panel are LR-HPV and lower panel are HR-HPV sequences). The orange box shows the PDZ-binding motifs in E6 of HR-HPV. (C) Schematic representation of the E6/E7 separation through the insertion in E6 and the proximity of E6 stop and E7 starts in HR-HPV.
The discriminating region identified in the E6 gene contains an insert with a conserved sequence in all HR-HPV strains. The insert contains an in-frame stop codon for the E6 coding sequence, which eliminates the overlap between the coding sequences of E6 and E7 that is characteristic of the LR-HPV genomes, but results in nearly identical lengths of the E6 proteins in HR and LR-HPV strains albeit with unrelated C-terminal sequences of 8-19 amino acids (Figure 2B). The unique C-terminal sequence of HR-HPV E6 that originates from the insert contains a PDZ domain-binding motif X-T-X-V/L at the very C-terminus of E6 in almost all HR-HPVs. Indeed, several PDZ domain-containing proteins have been identified as binding partners of HR-E6, including hDlg, hScrib, MAGI-1, MAGI-2, MAGI-3, and MUPP126,29,30,33–35. These interactions that are unique for HR-HPV are likely to be a key contributing factor to HR-HPV induced oncogenesis36.
We observed that the sequence similarity between the insert sequences among HR-HPV strains is more pronounced at the nucleotide level than at the amino acid level (See METHODS for details and Extended data: Figure S331). Combined with the separation between the coding regions of E6 and E7 resulting from the insertion, and the proximity of E6 stop codon to the E7 start codon, this finding led us to hypothesize that the insert has an additional role as a regulatory region. Furthermore, as E7 has been previously identified as the dominant oncogene37, the lack of genomic determinants of HR-HPV within the E7 gene is compatible with the possibility that the insert contains regulatory elements enhancing E7 expression in HR-HPV strains.
In HR-HPV strains, E6 and E7 proteins are translated from a polycistronic pre-mRNA38, where translation reinitiation has been suggested as mechanism39–41. However, the close proximity of the E6 stop codon to the E7 start codon in HR-HPV (only a few nucleotides separating these codons) could inhibit re-initiation39,40. Therefore, it has been suggested that ribosomal reinitiation is enabled through the E6*I splice variant in which the intercistronic distance between the translation termination codon of E6*I and the E7 initiation codon is increased41. However, several studies have reported that E7 translation is independent of splicing within the E6 open reading frame40,42,43, undermining this interpretation.
Several cases of coupled termination-reinitiation for polycistronic mRNA with proximate stop and start codons are evident for translation of eukaryotic virus genes44–51. The efficiency of this process depends on the close proximity of the termination and reinitiation sites51,52, and the presence of motifs complementary to the 18S rRNA in the mRNA sequence45,46,51,53. Given the proximity of the E6 termination site to the E7 initiation site codon that results from the insertion in the HR-HPV strains conferred by the insert, we investigated the possibility of coupled termination-reinitiaion of E6 and E7 in these strains. Notably, within the inserted sequence in the vicinity of the E7 start codon, we identified two conserved regions that are complementary to the sequences in 18S rRNA hairpins 26 and 27 which are commonly involved in the interactions between ribosomes and virus IRES54 (Figure 3A). The first region of complementarity consistently forms an internal loop and a relaxed, unpaired structure in the predicted optimal and sub-optimal E6-E7 mRNA foldings of HR-HPV strains54 (Extended data: Figure S431, see Methods). The second region of 18S RNA complementarity overlaps with the E7 start site, and hence might function independently in the re-initiation of E7 translation (Figure 3B). These findings suggest that the insertion could enable coupled termination-reinitiation of E6 and E7 proteins, enhancing their combined expression in HR-HPV, and thus promoting the oncogenic transformation induced by these viruses.
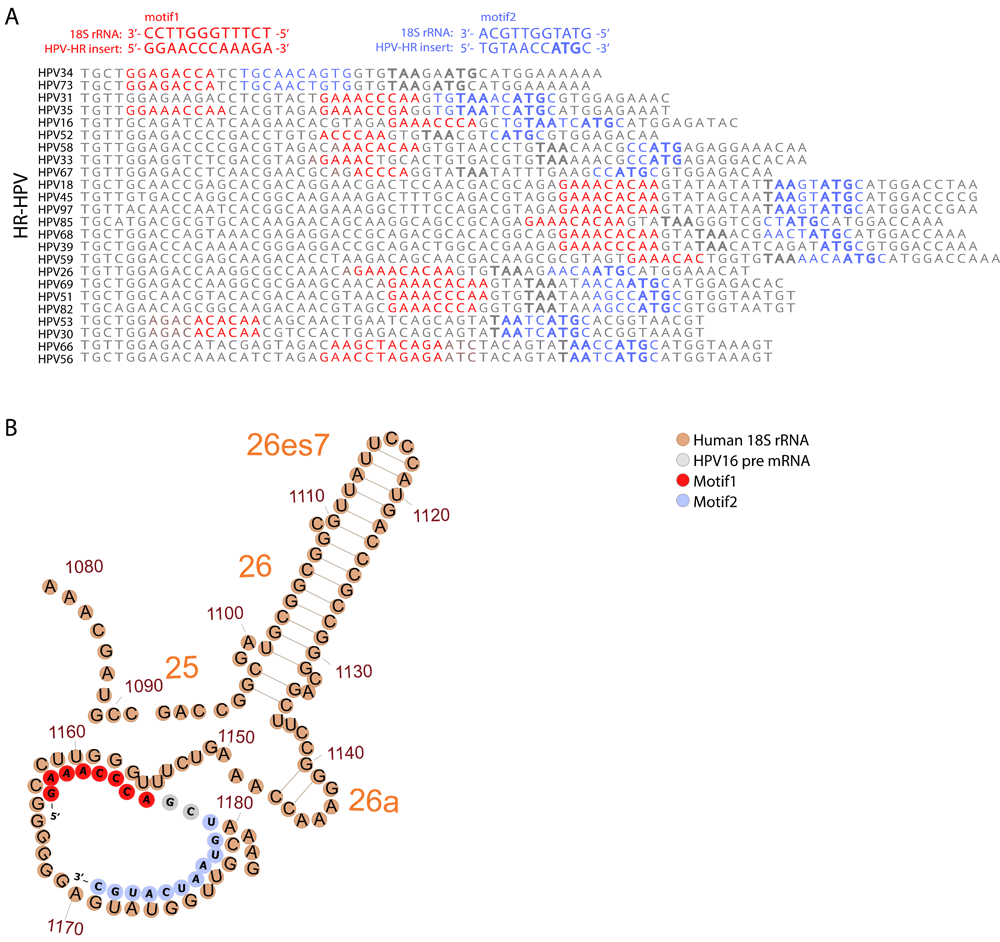
(A) Comparison of 18S rRNA complementary sequences for different HR-HPV strains. The distal and proximal to the E7 start motifs (motifs 1 and 2) to the E7 start codon are shown in red and blue, respectively. The E6 stop codons (TAA) and E7 start codons are marked in bold. (B) An illustration of the potential interactions between folded HPV16 pre-mRNA (grey, red and blue) and the human 18S rRNA (brown). The two motifs are marked on the HPV-structure in red (motif 1) and blue (motif 2).
The genus Alphapapillomavirus includes HPV types that are uniquely pathogenic. However, the events in the HPV genome evolution that led to the carcinogenic potential of some alpha-HPV types remain poorly understood. Here, we revisited this problem by performing a search for genomic determinants of the oncogenic risk of alpha-HPV types and identified a unique insert in the 3’-terminal region of the E6 oncoprotein gene in all HR HPV strains. To the best of our knowledge, this insert in HR-HPV genomes has not been reported previously. The insertion maintains closely similar lengths of E6 proteins in HR-HPV and LR-HPV types, which could explain why it has been overlooked in previous HPV genome analyses.
We hypothesize that the insertion makes a dual contribution to the oncogenicity of the HR-HPV types. First, the inserted sequence changes the C-terminal amino acid sequence of E6 and creates a PDZ domain-binding motif that is unique to HR-HPV types. The experimentally demonstrated interaction between the E6 proteins of HR-HPV and several PDZ domain-containing proteins is thought to make a substantial contribution to HPV-induced tumorigenesis27,29,55. Interestingly, PDZ-binding motifs have been identified also in several other oncogenic viruses, such as HTLV-1, adenovirus RhPV1 and beta-HPV856,57. Second, the insert eliminates the overlap between the E6 and E7 coding regions, implying a possible role as a regulatory region. We find that almost all HR-HPV genomes contain the sequence T-A/G-T-A-A-T/A in the insert near the end of the E6 coding sequence, a closely similar sequence to that functioning as the early promoter at the 5’ end of E6 and is employed for producing the mRNAs for both E6 and E758,59. However, in HR-HPV strains, unlike the case of the LR-HPV39, there are no reports of an independent E7 promoter, and E6 and E7 are both translated from a polycistronic mRNA.
Further investigating the potential regulatory role of the inserted sequence, we identified two conserved motifs that are complementary to the human 18S rRNA; interaction of such motifs with the rRNA has been shown to play a role in coupled termination-reinitiation for several viral genes45,46,51,53. The first motif forms an internal loop in the predicted mRNA secondary structure of E6E7, whereas the second one overlaps with the E7 initiation codon. Given the evolutionary conservation of these motifs and the close proximity of E6 termination site to the E7 initiation site, it appears plausible that coupled termination-reinitiation promoted by the insert sequence is a central mechanism for E7 translation in HR-HPV strains. Given the lack of additional major genomic determinants that would consistently differentiate between HR-HPV and LR-HPV, it seems likely that the insert in E6 is the primary cause behind the emergence of oncogenic HPV.
Multiple alignments of nucleotide and amino acid sequences that were obtained from GenBank (Extended data: Table S131) were generated using MAFFT v7.40760 with default parameters. Maximum likelihood phylogenetic analysis was performed using the resulting alignments and PhyML 3.1 software61, with the Bayesian information criterion, NNI tree improvement and an LRT SH-like likelihood method for support estimation.
To apply Support Vector Machines (SVM62, using Matlab 2018b fitsvm function) to the nucleotide sequences, we first encoded the data with numbers, where each nucleotide is coded as ‘1’ and each gap as ‘0’. We searched for alignment regions with deletions or insertions in multiple HPV strains that are surrounded by high confidence alignment regions (alignment regions of length > 15 bp containing less than 5% of gaps in each position) because these are most likely to contain relevant differences within conserved genomic regions. Within these regions, we then trained the SVM to classify alpha-HPV strains based on their oncogenic potential. The performance of the SVM was evaluated by leave-one-out cross validation, and regions with the overall balanced accuracy >0.75 (the average of the accuracy for positive and negative classes) were selected for further analysis.
To apply SVM (using Matlab 2018b fitsvm function) to the amino acid sequences of E6 and E7 proteins, we first encoded the amino acids with numbers using the frequencies of each amino acid in each protein and the BLOSUM62 matrix32. For each position, the most frequent amino acid was identified, and the amino acids in each protein were encoded by their BLOSUM62 distances from the most frequent amino acid in the respective position. We then trained the SVM to classify alpha-HPV strains based on their oncogenic potential using the coded protein sequences. Positions surrounded by high-confidence alignment regions (length > 5 amino acids and containing less than 5% of gaps in each position) were selected for further analysis. For these positions, we evaluated the performance by leave-one-out cross validation, and regions with the overall balanced accuracy >0.75 were selected for further estimation of significance using a permutation test.
To estimate the significance of the identified regions, i.e. to determine whether similar differences could be observed by chance, we performed a permutation test (Extended data63), controlling for the topology of the reconstructed phylogenetic trees. To this end, the labels were randomly permuted while maintaining unified labels for clades with high similarity. For each identified region, the likelihood of obtaining an equivalent or higher performance for the length of the region within the respective protein was evaluated by calculating an empirical P-value. We consider regions with permutation P-value <0.05.
The most stable secondary structures were predicted for all HR-HPV strains and their free energy values were calculated using the Vienna package54. Afold and Mfold were applied for prediction of optimal and sub-optimal mRNA structures64,65. Target opening free energy was estimated for motifs 1 and 2 using optimal and sub-optimal RNA structures, as described previously66. The sequence fold variants with the lowest secondary-structure free energy are presented in the Extended data: Figure S431. The formation of intermolecular mRNA-rRNA duplexes and hybridization affinity of the E6-E7 inserts to ribosomal RNA were evaluated using the Hybrid software with default parameters, with a ΔG threshold of ≤−10 kcal/mol67. The human 18S rRNA 2D structure was extracted from the structures of X-ray structure of eukaryotic ribosome (http://apollo.chemistry.gatech.edu/RibosomeGallery H%20sapiens/SSU/index.html; 68,69).
Nucleotide sequences of HPV and amino acid sequences of HPV E6 and E7 proteins are available as extended data.
Harvard Dataverse: A unique insert in the genomes of in high-risk human papillomaviruses with a predicted dual role in conferring oncogenic risk, https://doi.org/10.7910/DVN/FUGEUB31.
This project contains the following extended data:
Table 1: Complete nucleotide sequences and the amino acid sequences of HPV E1, E2, E4, E5, E6, E7, L1 and L2 proteins. This is the only source data that was required and employed for the analysis reported in this work.
Figure S1: Maximum likelihood trees obtained with alignments of E6, E7, E1, E2, E4, E5, L1 and L2 amino acid sequences of alpha-HPV strains.
Figure S2: The balanced accuracy (y-axis) obtained from a leave-one-out cross validation for predicting risk category (HR vs LR) of alpha-HPV strains using BLOSUM62 coding of amino acid sequences, of different positions (x-axis) E6 (A) and E7 (B). Zero-accuracy was assigned to regions surrounded with low confidence alignment.
Figure S3: Boxplots showing the distributions of the identity fraction of each nucleotide (NN) and amino acid (AA) in the genome and protein sequences of the insertion (not considering gaps for both NN and AA). The individual identity fractions of each position are overlaid.
Figure S4: RNA fold secondary structure prediction of HR HPV strains 16 (A), 18 (B), 45 (C) and 31 (D). The nucleotides of the first motif are marked in red, and of the second motif in purple. E7 AUG is noted in red font.
Data are available under the terms of the Creative Commons Zero “No rights reserved” data waiver (CC0 1.0 Public domain dedication).
Zenodo: Permutation test controlling for HPV strains, http://doi.org/10.5281/zenodo.324223163. Apache License, Version 2.0.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)