Keywords
differential gene expression analysis, gene set analysis, enrichment analysis, network analyis, GSEA
This article is included in the RPackage gateway.
This article is included in the Bioconductor gateway.
differential gene expression analysis, gene set analysis, enrichment analysis, network analyis, GSEA
This revised manuscript is based upon valuable input from the reviewers. Most importantly, installation and loading of required packages is now taken care of.
See the author's detailed response to the review by Jill P. Mesirov and Alexander Wenzel
See the author's detailed response to the review by Rita Casadio
See the author's detailed response to the review by Kimberly Glass
See the author's detailed response to the review by Monther Alhamdoosh
Interpretation of whole-transcriptome differential expression studies is often difficult because the sheer volume of the differentially expressed genes (DEGs) can be overwhelming. It is common place in designed experiments with more than just a marginal biological effect to find several thousands of differentially expressed genes (DEGs). One way to handle the vast numbers and to identify the biological consequences of gene expression changes is to associate them with overarching processes involving a whole set of genes, such as Gene Ontology (GO) terms or Kyoto Enzyclopedia of Genes and Genomes (KEGG) pathways.
Curated genesets have been designed or discovered for a wide range of common contexts, such as, a biological process, molecular pathway, or tissue localization1,2. They have been introduced in the past not only to reduce complexity and to improve interpretability but also to increase statistical power by reducing the number of performed tests. As it turns out, this often results in finding hundreds of differentially regulated pathways1.
As with co-expressed genes, many of the pathways exhibit strong mutual correlation because they contain a large proportion of shared genes which is in turn a result of the fact that many of them describe closely related aspects of an overarching biological theme. Therefore, to further increase interpretability of differential geneset regulation and to capture the global change of a biological phenotype, it would be desirable to identify possibly existing umbrella organizations among genesets.
Networks are ideal to model dependencies, interactions, and similarities among individuals3–5, be it people, computers, genes, or genesets. The degree of connectivity between them can have an influence on information flow and defines communities or cliques, i.e., clusters of highly connected nodes within and infrequent connections between them.
In order to construct a geneset network, a similarity measure is required and can be defined as the fraction of common genes, also called the Jaccard index6. Other ways to measure similarity among genesets include, for instance, coexpression strength as implemented in WGCNA7,8.
Community detection based on network topology is a standard problem in the analysis of social networks9,10. Well-established algorithms allow for computationally efficient clustering of genesets and can be used to identify highly connected sub-networks. There is no unique or optimal method available but many options exist. Popular methods to define clusters include the edge-betweenness criterion, the Infomap or the Louvain algorithm (igraph), as well as hierarchical or kmeans clustering.
Once geneset clusters are defined they can be characterized by their size and connectivity and thus prioritized and ranked. In particular, the clusters can be categorized as singletons, doublets, medium and large or dense and loose clusters.
Network analysis not only allows for detection of clusters and performance of measurements on them, networks are also straightforward and appealing visualizations of similarities among genesets. There are a couple of interactive visualization software tools available, of which Cytoscape is probably the most popular11. In some cases interactivity is useful but the emphasis here is to provide some of Cytoscape’s features without any human intervention for easy integration into automatic analysis pipelines. For instance, automatic labeling of communities using the n most frequent terms was adopted here, similar as in Kucera et al.12.
The purpose of this step-by-step workflow is to provide a fully automated and reproducible procedure for downstream analysis and visualization of differential geneset analysis results in R13. The focus is on supporting scientists in result interpretation by bringing order into the list of differentially regulated genesets based on biological rather than pure statistical arguments. The workflow is suitable for any kind of geneset library including new or custom sets and any kind of geneset analysis method.
Starting with differential expression analysis of a model dataset, geneset analysis is performed based on the MSigDB library. A geneset network is constructed to identify isolated genesets (singletons) and geneset pairs (doublets). Larger connected sub-networks are then split into smaller clusters of closely related genesets describing similar processes. The effect of each modification step on the network topology is visually documented in Figure 1–Figure 4. Using the most frequently occurring terms in the geneset names of a cluster, an attempt to automatically assign cluster labels is made. Finally, all labeled clusters of genesets are plotted to provide a one page overview of the results.
The packages required for this workflow provide plotting functions (ggplot2 and relatives), network functions (igraph14, etc.), text analytics functions (wordcloud, etc.) and gene expression analysis functions DESeq215, limma16, and org.Hs.eg.db. The Rmarkdown version of this article further requires the Bioconductor package BiocWorkflowTools to be installed.
cranpcks = c("ggplot2", "gplots", "cowplot", "RColorBrewer", "knitr", "ggrepel", "reshape2", "kableExtra","igraph", "GGally", "network", "sna", "intergraph", "wordcloud", "tm", "SnowballC", "BiocManager") biocpcks = c("DESeq2", "limma", "org.Hs.eg.db", "airway") tmp = sapply(cranpcks, require, character.only=T, warn.conflicts=F, quietly=T) toinstall = names(tmp)[!tmp] if(length(toinstall)>0){ install.packages(toinstall, quiet=T) sapply(toinstall, require, character.only=T, warn.conflicts=F, quietly=T) } tmp = sapply(biocpcks, require, character.only=T, warn.conflicts=F, quietly=T) toinstall = names(tmp)[!tmp] if(length(toinstall)>0) { BiocManager::install(toinstall) sapply(toinstall, require, character.only=T, warn.conflicts=F, quietly=T) }
In addition to and often based on igraph, several R packages for network visualization are available and described in the form of tutorials17,18.
We are using the popular airway data set19 and perform a simple differential expression analysis.
data(airway) dds = DESeqDataSetFromMatrix(countData = assay(airway), colData = colData(airway), design = ~ cell + dex) dds$dex = relevel(dds$dex, "untrt") dds = DESeq(dds, betaPrior = T) res = results(dds, contrast = c("dex", "trt", "untrt"))
We are using the popular org.Hs.eg.db package based on the UCSC annotation database and keep only genes with a unique mapping.
res$entrezgene = unname(mapIds(org.Hs.eg.db, keys = rownames(res), column = "ENTREZID", keytype = "ENSEMBL")) res = subset(res, subset = !is.na(res$entrezgene) & !is.na(res$stat)) res = res[-which(duplicated(res$entrezgene)), ]
We are using the popular KEGG, Reactome, and Biocarta pathways from the MSigDB gene set library C2. The following chunk guarantees that the gene set library list object is called gset.
url = "http://bioinf.wehi.edu.au/software/MSigDB/human_c2_v5p2.rdata" temp.space = new.env() bar = load(url(url), temp.space) gset = get(bar, temp.space) rm(temp.space) gs.libs = sapply(names(gset), function(x) strsplit(x, "_")[[1]][1]) gset = gset[which(gs.libs %in% c("KEGG", "REACTOME", "BIOCARTA"))] idx = ids2indices(gene.sets = gset, identifiers = res$entrezgene)
Alternatively, the same result can be obtained using the EGSEAdata experiment package19b.
library(EGSEAdata)
library(EGSEA)
gset = EGSEA::buildMSigDBIdx(entrezIDs = res$entrezgene,
species = "human",
geneSets = "c2", min.size = 3)
idx = gset$c2@idx
gs.libs = sapply(names(idx), function(x) strsplit(x, "_")[[1]][1])
idx = idx[which(gs.libs %in% c("KEGG", "REACTOME", "BIOCARTA"))]Competitive gene set enrichment analysis is performed using the function camera() from the limma package. We include uni-directional and bi-directional enrichment by using both the test statistics (“up” or “down”) and its modulus (“mixed”) for gene set testing. We limit the following network analysis to gene sets with a FDR < 0.05.
dat = cameraPR(res$stat, idx, sort = F) dat$PValue.Mixed = cameraPR(abs(res$stat), idx, sort = F)$PValue dat$FDR.Mixed = p.adjust(dat$PValue.Mixed, method = "BH") dat$name = rownames(dat) dat$Direction = as.character(dat$Direction) dat$Direction[dat$FDR > 0.05] = "Mixed" dat$Direction[dat$Direction == "Mixed" & dat$FDR.Mixed > 0.05] = "NOT" dat$Direction = factor(dat$Direction, levels=c("NOT", "Up", "Down", "Mixed")) idx = which(dat$Direction == "Mixed") if(length(idx) > 0) dat$FDR[idx] = dat$FDR.Mixed[idx] dat = dat[, -grep("\\.Mixed", names(dat))] dat = dat[dat$Direction != "NOT", ] dat$Direction = factor(dat$Direction, levels=c("Up", "Down", "Mixed"))
Starting from 1077 gene sets, 264 are found to be differentially regulated. Many of them are expected to describe similar processes and to be highly correlated.
We construct a gene set network based on the proportion of common genes as the inverse distance measure. The nodes are gene sets which are connected by edges if the Jaccard index
is larger than a preset threshold, J > 0.2. While this threshold is somewhat arbitrary it has proven to be a reasonable one in many projects. As a guide for finding a reasonable threshold a broad distribution of disjoint cluster sizes is desired. Network analysis does not help if the cutoff is too large (no connections) or too small (all sets are connected with each other). In any case, it is strongly recommended to investigate its effect on the quality of the results.
# only keep gene sets present in the data id.keep = which(names(gset) %in% dat$name) gset = gset[id.keep] # adjacency matrix m.adj = sapply(gset, function(x) sapply(gset, function(y) length(intersect(unlist(x), unlist(y) )) ) ) diag(m.adj) = 0 # Jaccard index matrix NGenes = sapply(gset, length) m.union = outer(NGenes, NGenes, "+") - m.adj m.jacc = m.adj / m.union
The Jaccard matrix, or adjacency matrix, can be conveniently used to construct a network object using the function igraph::graph_from_adjacency_matrix(). In this example geneset, similarity is measured using all member genes irrespective of whether they were detected and present in the data. Alternatively, one could include only genes present in the data depending on whether the current data seem more relevant and trustworthy or the prior information given by the geneset definition. Graphical display is achieved here using ggnet::ggnet2() (Figure 1).
# choose node colors palette = brewer.pal(9, "Set1")[c(1,2,9)] names(palette) = c("Up", "Down", "Mixed") # apply cutoff to Jaccard matrix m.adj1 = m.adj * (m.jacc > 0.2) # construct network object net = graph_from_adjacency_matrix(m.adj1, "upper", diag = F, weighted = T) # add vertex features V(net)$size = dat$NGenes V(net)$color = palette[dat$Direction] V(net)$Direction = as.character(dat$Direction) # plot ggnet2(net, size = 2, color = "Direction", palette = palette, edge.size = 1, edge.color = "#99CC33")
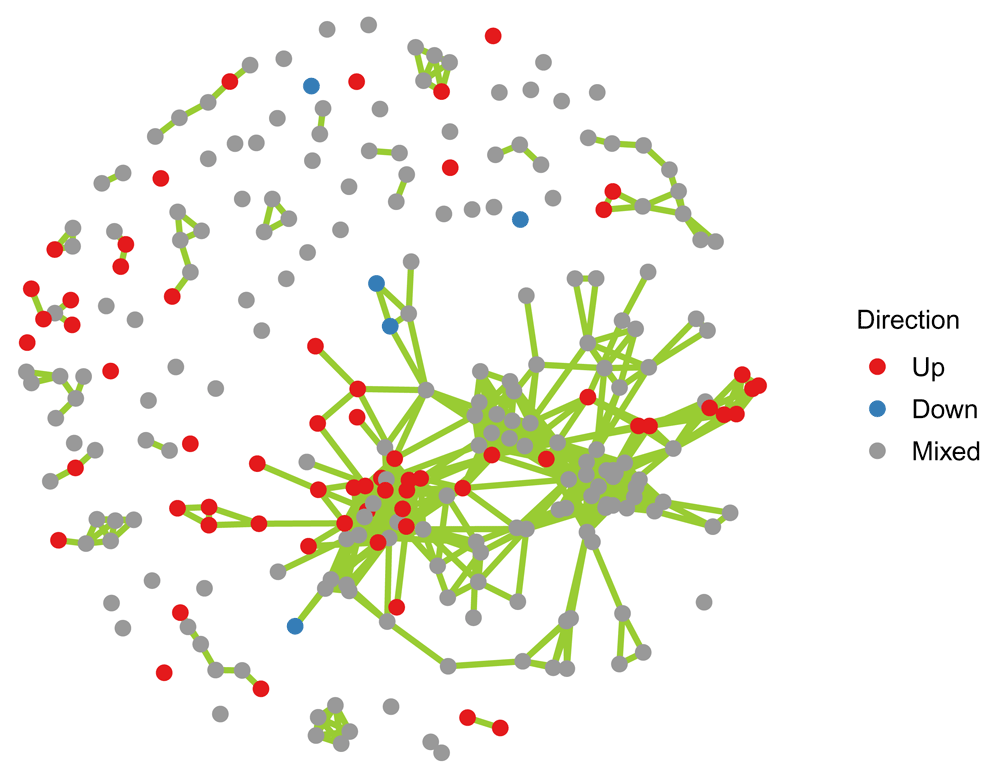
Node colors indicate whether the member genes of a set are predominantly up or down regulated or whether there is no preferential direction (mixed).
In the following, components of the network for which network analysis does not improve interpretability are identified and put to aside. This includes singletons, i.e., genesets not connected to any other geneset, and doublets, also termed binary systems or dumbbells, i.e., pairs of genesets connected with each other but isolated from the rest.
singletons = which(igraph::degree(net) == 0) net1 = delete_vertices(net, singletons) in.single = which(dat$name %in% V(net)$name[singletons]) tab = dat[in.single, ] tab$FDR = signif(tab$FDR, 2) tab$name = gsub("_", " ", tab$name) tab = kable(tab[,c("name", "NGenes", "Direction", "FDR")], row.names = F, format = "latex", caption = "List of all singletons, i.e., genesets without sufficient overlap with any other geneset.") kable_styling(tab, latex_options = "scale_down", font_size = 8)
In total, 49 singletons were identified and excluded from further analysis (Table 1). It is important to note that these genesets, while down-prioritized for the time being, may still be worthwhile investigating later.
ggnet2(net1, size = "size", max_size = 4, color = palette[V(net1)$Direction], size.cut = 4, edge.size = 1, edge.color = "#99CC33")
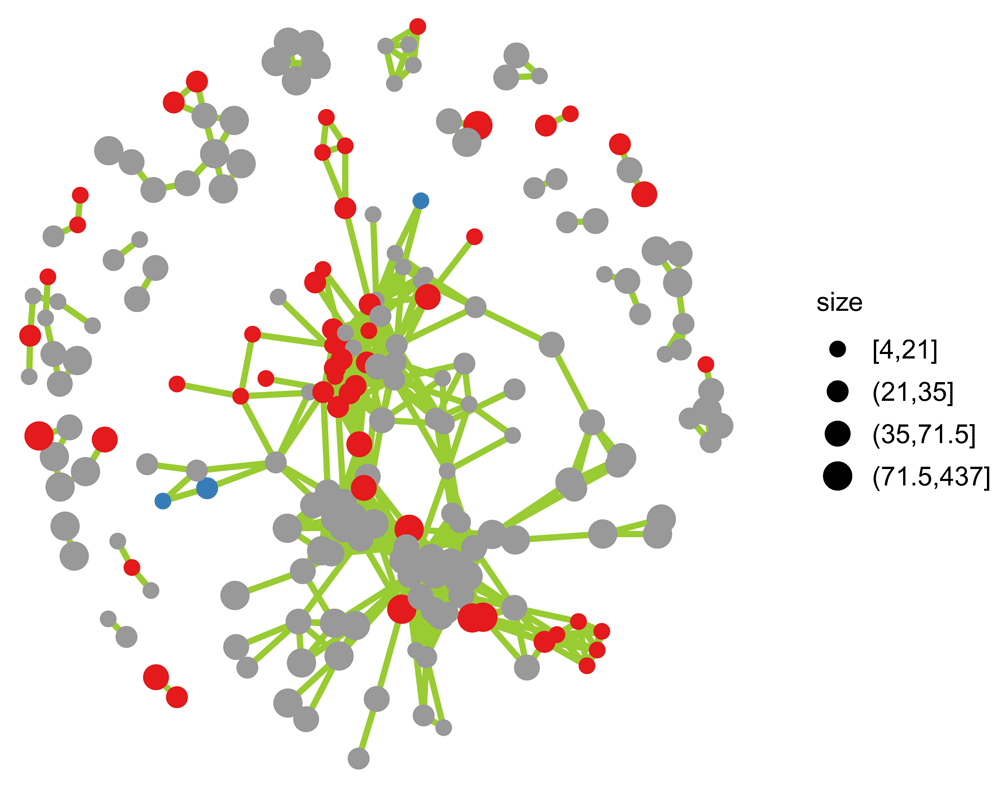
The color scheme is the same as above. The node size corresponds to the number of genes in a set.
Figure 2 shows the remaining network clusters, with the size of the nodes representing the number of genes in the set.
Next we also want to separate clusters with less than 3 gene sets. To do so, we separate disjoint subnets as individual objects, count their members, and delete all vertices belonging to clusters of size smaller than 3.
clu1 = igraph::components(net1) clu.lt3 = which(sizes(clu1) < 3) v.clu.lt3 = which(clu1$membership %in% clu.lt3) net2 = delete_vertices(net1, v.clu.lt3) clu2 = igraph::components(net2) in.clu.lt3 = which(dat$name %in% V(net1)$name[v.clu.lt3]) tab = dat[in.clu.lt3, ] tab$FDR = signif(tab$FDR,2) cludp = clu1$membership[v.clu.lt3] cludp = data.frame(name = names(cludp), id = as.numeric(cludp)) tab = merge(tab,cludp) tab$name = gsub("_", " ", tab$name) tab = kable(tab[order(tab$id), c("id", "name", "NGenes", "Direction", "FDR")], row.names=F, format = "latex", caption = "List of binary clusters as indicated by the id column.") kable_styling(tab, latex_options = "scale_down", font_size = 8)
In Table 2, consecutively listed gene sets with the same id belong to the same binary cluster. Often these are gene sets from different libraries describing the same biological process or phenotype. In total, 16 binary clusters were identified, for which network analysis would not be useful.
set.seed(16) nodecol = colorRampPalette(brewer.pal(9, "Set1")[sample(9)])(max(clu2$membership)) ggnet2(net2, size = "size", max_size = 4, color = nodecol[clu2$membership], size.cut = 4, edge.size = 1, edge.color = "grey")
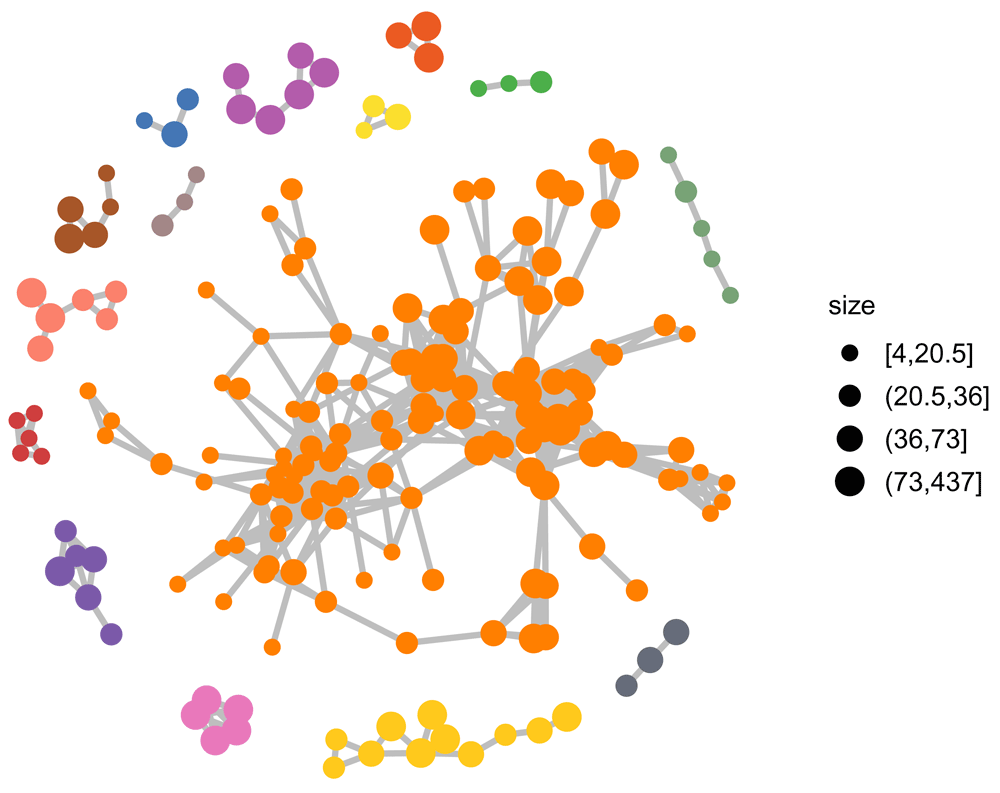
Colored according to disjoint subnetworks.
Without singletons and binary clusters, we are left with larger disjoint subnets (Figure 3).
The larger disjoint clusters may consist of so-called communities, i.e., sub-networks of highly inter-connected nodes that stick together by only one or a few edges. We are using the popular edge betweenness property to identify these community-connecting edges and remove them in order to split large clusters into smaller ones.
net2 = delete_edge_attr(net2, "weight") clu3 = cluster_edge_betweenness(net2) # delete edges between communities net3 = delete_edges(net2, which(as.vector(crossing(clu3, net2))) ) # remove clusters of size <3 small_cluster_ids = which(sizes(clu3) < 3) small_cl_v = which(clu3$membership %in% small_cluster_ids) net3 = delete_vertices(net3, small_cl_v) clu3 = igraph::components(net3) nodecol = c(brewer.pal(9, "Paired"), brewer.pal(9, "Set3") ) nodecol = colorRampPalette(nodecol)(max(clu3$membership)) ggnet2(net3, size = 0, color = nodecol[clu3$membership], edge.size = 1.0, edge.color = "grey") + geom_point(size = 2, color = "black") + geom_point(aes(color = color), size = 1)
The result of this network-based clustering is shown in Figure 4

In analogy to the popular interactive network visualization tool cytoscape12, we attempt to generate automatic labels for gene set clusters. Gene set names are split into individual words and counted within each cluster. The four most frequent terms occurring at least twice are used as labels. The function clust_head() is defined for this purpose and contains an exclusion list of words not used.
t.rW = c("cell", "process", "regulation", "negative", "positive", "signaling", "response", "stimulus", "signal", "activity", "protein", "involved", "component", "level", "effector", "event", "projection", "organismal", "cellular", "modification", "pathway", "mediated", "dependent", "organization", "group", "target", "biocarta", "kegg", "reactome") clust_head = function(x){ txt = unlist(strsplit(x, "_")) txt = Corpus(VectorSource(txt)) txt = tm_map(txt, PlainTextDocument) txt = tm_map(txt, removePunctuation) txt = tm_map(txt, removeNumbers) txt = tm_map(txt, content_transformer(tolower)) txt = tm_map(txt, removeWords, c(t.rW, stopwords("english"))) tdm = TermDocumentMatrix(txt) m = as.matrix(tdm) word_freqs = sort(rowSums(m), decreasing=TRUE) word_freqs = word_freqs[word_freqs>1] word_freqs = paste(names(word_freqs)[1:4], collapse=" ") gsub("[[:space:]]?NA[[:space:]]?", "", word_freqs) }
There are many possibilities to visualize geneset clusters and often a compromise between information content and crowding has to be found. Here, we are producing a lattice of network plots, one for each sub-net, with the automatic annotation as title (Figure 5). We begin by generating the cluster titles using the clust_head() function followed by cleaning up and ordering by cluster size.
clust = data.frame(cl = clu3$membership) rownames(clust) = names(V(net3)) # generate cluster titles cl3.lab.txt = as.character(tapply(rownames(clust), clust$cl, clust_head)) # remove NAs cl3.lab.txt = gsub("[[:space:]]?NA[[:space:]]?", "", cl3.lab.txt) clu3 = igraph::components(net3) clu.order = order(clu3$csize, decreasing = T) clu3$mem = match(clu3$membership, clu.order)
Then we generate a list of ggplot objects, one for each cluster or sub-net. For smaller sub-nets, the nodes are labelled with the first 4 words of their names; the first word was removed before as it is usually the name of the geneset library. For larger sub-nets, this is not feasible without overprinting. Titles are missing if none of the words from the geneset names occurred more than once. This may be indicative for a semantically mixed cluster or for sparse prior knowledge.
# generate a list of ggplots g = list(max(clu3$membership)) set.seed(7042016) for (ii in 1:max(clu3$membership)) { subgf = induced_subgraph(net3, which(clu3$mem == ii)) # generate titles with one optional line break title = substr(toupper(cl3.lab.txt[clu.order][ii]), 1, 60) if (nchar(title) > 25) { title = sub("(^.{10,30})[[:space:]]","\\1\\\n", title) } # generate node labels using word 2-5 of the geneset name v.label = names(V(subgf)) v.label = lapply(v.label, function(x) strsplit(x, "_")[[1]]) v.label = sapply(v.label, function(x) paste(x[2:min(5, length(x))], collapse = "_")) # clean up geneset names v.label = gsub("_PATHWAY","", v.label) v.label = gsub("_SIGNALING", "", v.label) # introduce line breaks v.label = gsub("_","\n", v.label) # remove node labels for large clusters if (length(v.label) > 5) v.label = rep(NA, length(v.label)) g[[ii]] = ggnet2(subgf, edge.size = 1, edge.color = "#99CC33", label = F, size=V(subgf)$size, max_size = 3, size.cut = 4, color = palette[V(subgf)$Direction]) + theme(legend.position="none", plot.title = element_text(size=6), panel.grid = element_blank()) + geom_label_repel(label = v.label, size=1.2, box.padding = 0.1, label.padding = 0.1) + ggtitle(title) }
nr.cols = min(4,max(clu3$membership)) nr.rows = ceiling(max(clu3$membership) / nr.cols) width = sapply(g, function(x) nrow(x$data)) grid.arrange = getFromNamespace("grid.arrange", asNamespace("gridExtra")) grid.arrange(grobs = g[seq(16)], ncol = nr.cols)
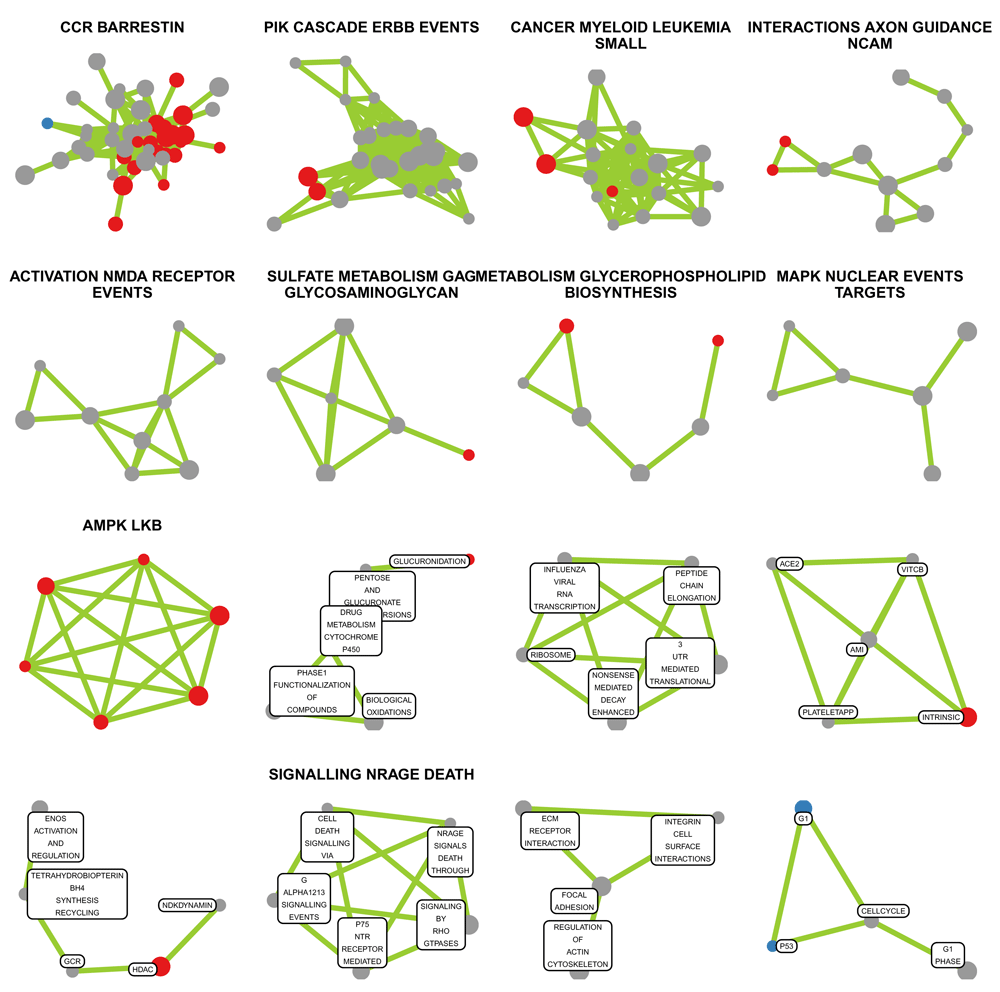
Only the first 16 connected subnets are shown. Geneset labels are omitted for clusters with more than 5 members.
We have presented an automated workflow based on a small number of R packages for prioritization and visualization of gene set analysis results using networks, which we call RICHNET. We demonstrated how community detection facilitates categorization of differentially regulated gene sets into singletons and clusters of different size ranges. Automated label generation allowed to associate these clusters with biological themes or processes of which the member gene sets are part of.
The RICHNET workflow could be altered or extended quite naturally in a number of ways but the version presented here is the one we typically apply in our research service projects. One advantage over other approaches is that it does not depend on a particular geneset library or geneset analysis method. Any means of selecting genesets of interest can be used. Specific hierarchically constructed genesets, such as GO terms, would offer a straightforward way to arrive at a more global process description using higher levels in their tree structure. A second advantage is that it does not depend on the existence of a good quality gene or protein interaction network for the particular organism or disease state which is often not feasible. Only very few genesets are network-based (e.g. KEGG pathways) and would thus offer a straight-forward way to use an a priori network topology. Thirdly, similar as in reference 8, a geneset similarity network could be constructed in the form of a co-enrichment network from GSVA enrichment scores20 using weighted co-expression network analysis (WGCNA)7. However, this approach relies on a relatively large sample size whereas the sample size requirement of RICHNET is not more than the GSA it relies on.
As an alternative to the networks of genesets described here, networks of genes could be created in a reciprocal way. The underlying similarity metric between genes could be defined as the proportion of common genesets among all genesets they are part of. This approach would be equivalent to a STRING-DB network with “databases” as the only interaction allowed21.
One possible future extension of the RICHNET workflow could be the introduction of a consensus similarity metric from multiple initial networks and different community detection or cluster algorithms to improve stability against noise. A second avenue forward could be the introduction of interactive graphics in 2D or 3D17 to allow moving, pulling, rotation or zoom and display of node specific or edge specific information.
Some may argue in favor of encapsulating the RICHNET workflow in an R or Bioconductor package. However, it is our strong believe that for the sake of transparency and given the straightforward nature of the code it serves better to publish it openly. This way we encourage the users to adapt it to their specific requirements, to improve and expand on it.
The data used in this workflow is included in the airway R-package19.
The R markdown file for this workflow is available at: https://doi.org/10.5281/zenodo.327156513.
License: Creative Commons CC BY license.
This workflow depends on various packages from version 3.7 of the Bioconductor project, running on R version 3.5.0 or higher. A complete list of the packages used for this workflow is shown below:
sessionInfo() R version 3.5.1 (2018-07-02) Platform: x86_64-w64-mingw32/x64 (64-bit) Running under: Windows 10 x64 (build 17134) Matrix products: default locale: [1] LC_COLLATE=English_United Kingdom.1252 [2] LC_CTYPE=English_United Kingdom.1252 [3] LC_MONETARY=English_United Kingdom.1252 [4] LC_NUMERIC=C [5] LC_TIME=English_United Kingdom.1252 attached base packages: [1] parallel stats4 stats graphics grDevices utils datasets [8] methods base other attached packages: [1] airway_1.2.0 SnowballC_0.5.1 [3] tm_0.7-5 NLP_0.2-0 [5] wordcloud_2.6 org.Hs.eg.db_3.7.0 [7] AnnotationDbi_1.44.0 limma_3.38.2 [9] DESeq2_1.22.1 SummarizedExperiment_1.12.0 [11] DelayedArray_0.8.0 BiocParallel_1.16.1 [13] matrixStats_0.54.0 Biobase_2.42.0 [15] GenomicRanges_1.34.0 GenomeInfoDb_1.18.1 [17] IRanges_2.16.0 S4Vectors_0.20.1 [19] BiocGenerics_0.28.0 igraph_1.2.2 [21] GGally_1.4.0 kableExtra_0.9.0 [23] knitr_1.20 reshape2_1.4.3 [25] ggrepel_0.8.0 cowplot_0.9.3 [27] gplots_3.0.1 ggplot2_3.1.0 [29] RColorBrewer_1.1-2 loaded via a namespace (and not attached): [1] colorspace_1.3-2 rprojroot_1.3-2 [3] htmlTable_1.12 XVector_0.22.0 [5] base64enc_0.1-3 fs_1.2.6 [7] rstudioapi_0.8 remotes_2.0.2 [9] bit64_0.9-7 xml2_1.2.0 [11] codetools_0.2-15 splines_3.5.1 [13] geneplotter_1.60.0 pkgload_1.0.2 [15] Formula_1.2-3 annotate_1.60.0 [17] cluster_2.0.7-1 intergraph_2.0-2 [19] readr_1.2.1 compiler_3.5.1 [21] httr_1.3.1 backports_1.1.2 [23] assertthat_0.2.0 Matrix_1.2-15 [25] lazyeval_0.2.1 cli_1.0.1 [27] acepack_1.4.1 htmltools_0.3.6 [29] prettyunits_1.0.2 tools_3.5.1 [31] bindrcpp_0.2.2 coda_0.19-2 [33] gtable_0.2.0 glue_1.3.0 [35] GenomeInfoDbData_1.2.0 dplyr_0.7.8 [37] BiocWorkflowTools_1.8.0 Rcpp_1.0.0 [39] slam_0.1-43 statnet.common_4.1.4 [41] gdata_2.18.0 xfun_0.4 [43] stringr_1.3.1 network_1.13.0.1 [45] ps_1.2.1 testthat_2.0.1 [47] rvest_0.3.2 gtools_3.8.1 [49] devtools_2.0.1 XML_3.98-1.16 [51] zlibbioc_1.28.0 scales_1.0.0 [53] hms_0.4.2 yaml_2.2.0 [55] memoise_1.1.0 gridExtra_2.3 [57] rpart_4.1-13 RSQLite_2.1.1 [59] reshape_0.8.8 latticeExtra_0.6-28 [61] stringi_1.2.4 genefilter_1.64.0 [63] desc_1.2.0 checkmate_1.8.5 [65] caTools_1.17.1.1 pkgbuild_1.0.2 [67] rlang_0.3.0.1 pkgconfig_2.0.2 [69] bitops_1.0-6 evaluate_0.12 [71] lattice_0.20-38 purrr_0.2.5 [73] bindr_0.1.1 htmlwidgets_1.3 [75] bit_1.1-14 processx_3.2.0 [77] tidyselect_0.2.5 plyr_1.8.4 [79] magrittr_1.5 bookdown_0.7 [81] R6_2.3.0 Hmisc_4.1-1 [83] sna_2.4 DBI_1.0.0 [85] pillar_1.3.0 foreign_0.8-71 [87] withr_2.1.2 survival_2.43-3 [89] RCurl_1.95-4.11 nnet_7.3-12 [91] tibble_1.4.2 crayon_1.3.4 [93] KernSmooth_2.23-15 rmarkdown_1.10 [95] usethis_1.4.0 locfit_1.5-9.1 [97] grid_3.5.1 data.table_1.11.8 [99] blob_1.1.1 callr_3.0.0 [101] git2r_0.23.0 digest_0.6.18 [103] xtable_1.8-3 munsell_0.5.0 [105] viridisLite_0.3.0 sessioninfo_1.1.1
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Thank you for this suggestion which is integrated in the revised manuscript.
Thank you for this suggestion which is integrated in the revised manuscript.
Thank you for this suggestion which is integrated in the revised manuscript.