Keywords
ChIP-seq, read mapping, peak calling, genomic region annotation, differential enrichment analysis, heatmaps, metaplots
This article is included in the RPackage gateway.
ChIP-seq, read mapping, peak calling, genomic region annotation, differential enrichment analysis, heatmaps, metaplots
bp: base pairs; ChIP-seq: chromatin immunoprecipitation followed by next-generation sequencing; cpm: counts per million; FDR: false discovery rate; GEO: Gene Expression Omnibus; GUI: graphical user interface; H3K36me3: histone H3 trimethylated at lysine 36; H3K4me3: histone H3 trimethylated at lysine 4; NCBI: National Center for Biotechnology Information; NGS: next-generation sequencing; PCR: polymerase chain reaction; PP: posterior probability; TMM: trimmed means of M values; TSS: transcription start site; UCSC: University of California Santa Cruz
Interactions between nuclear proteins and DNA are vital for cell and organism function. They control DNA replication and repair, safeguard genome stability, and regulate chromosome segregation and gene expression. Chromatin immunoprecipitation coupled with high-throughput sequencing (ChIP-seq) is a powerful method for assessing such interactions and has been widely used in recent years to map the location of post-translationally modified histones, transcription factors, chromatin modifiers and other non-histone DNA-associated proteins in a genome-wide manner. This progress has been fostered by the increasing technical feasibility and affordability of this technology, with more documentation and technical support available to researchers, including commercial library preparation kits, aided by the plummeting costs of sequencing and the ease of multiplexing samples. In light of this progress, ChIP-seq data sets are continuously deposited in publicly-accessible databases, such as the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) and the ENCODE consortium portal1. Therefore, there is an unprecedented wealth of epigenomic data in the public domain that can be used for integrative and correlative analyses.
This important advancement has not been accompanied with similar progress in user-friendly post-sequencing data analysis pipelines, which is still a significant bottleneck often handled by skilled bioinformaticians. A key step in ChIP-seq data analysis is to map reads to a reference genome assembly. Programs such as BWA2 or Bowtie/Bowtie23,4 are frequently employed for this purpose. Aligned data are then processed to find regions of enrichment along the genome, thereby identifying potential loci of DNA binding by the target protein or of deposition of the histone post-translational modifications of interest. This process is known as peak calling and can be performed by using algorithms such as MACS/MACS25,6 and SICER7. SAMtools is another popular software used in next-generation sequencing (NSG) analysis that provides various utilities for manipulating alignments, including sorting, merging, indexing and generating alignments in a per-position format8.
Following the initial and common steps in ChIP-seq data analysis mentioned above, downstream analysis is often more customizable and may present a hurdle, especially in the case of comparing data derived from different conditions or experimental settings. Artifacts arising from bias of DNA fragmentation, variation of immunoprecipitation efficiency, as well as polymerase chain reaction (PCR) amplification and sequencing depth bias, result in ChIP-seq experiments with distinct signal-to-noise ratios and impose great challenges to the computational analysis9. Several methods addressing the differential enrichment analysis problem, i.e. the detection of genomic regions with changes in ChIP-seq profiles between two distinct samples or sets of replicate samples, have been proposed10–12. Generally, these methods rely on the initial detection of candidate peak regions by a conventional peak calling algorithm, and then this peak-defining information is applied to analysis with methods tailored for the differential expression analysis of RNA-seq data such as edgeR13 or DESeq14. Downstream analysis of ChIP-seq data may also involve the annotation of genomic regions based on reference coordinates (e.g. distance from nearest transcription start site) and visualization of normalized coverage by means of heatmaps and metaplots.
Importantly, ChIP-seq data analysis typically relies, at least in part, on tools that have been designed to run primarily on Linux/Unix-based systems, while biologists who need to work with NGS may be unfamiliar with such operating systems. In the past few years, several packages designed to handle NGS data have been released for R, a popular platform-independent programming language and software environment for computing and graphics. Here we present ChIPdig15, an open-source application that leverages on several R packages to enable comprehensive and modular analysis of multi-sample ChIP-seq data sets through a user-friendly graphical user interface (GUI), allowing any experimental biologist with minimal computational expertise to use it easily.
ChIPdig15 is developed in R using package Shiny and relies on multiple R/Bioconductor packages, including QuasR and BSgenome16, for mapping reads to a reference genome assembly17, BayesPeak for peak calling18, csaw12 and edgeR13 for differential enrichment analysis, ChIPseeker for annotation of genomic regions of interest19, and EnrichedHeatmap for generating coverage heatmaps20. Packages used in several analysis modules implemented in ChIPdig are GenomicRanges21, valr22, shinyFiles, GenomicFeatures, ggplot2, ggsignif, reshape2 and circlize. The GUI allows integrative and interactive usage of these powerful libraries without requiring programming or statistical experience from the user. The required packages may be installed manually by the user or automatically upon launching the tool for the first time. A step-by-step tutorial on how to use ChIPdig is provided at https://github.com/rmesse/ChIPdig.
R and RStudio versions 3.4.1 and 1.0.44 or higher, respectively, are required, and both memory usage and execution times are contingent on the specificities of the intended analysis (e.g. size of input files, sequencing depths and size of the reference genome assembly).
ChIPdig has the following capabilities: (1) alignment of reads to a reference genome; (2) normalization and comparison of distinct ChIP-seq data sets; (3) annotation of genomic regions; (4) generation of comparative heatmaps and metaplots for visualization of normalized coverage in user-defined specific regions. Each capability corresponds to a specific analysis module which can be loaded separate from other modules and using different input files. For the alignment and annotation modules, the tool automatically fetches the desired reference assembly and gene model, respectively, from public repositories. Analysis of mammalian genome data is supported.
An earlier version of this article can be found on bioRxiv (DOI: https://doi.org/10.1101/220079).
To illustrate each module of the tool, raw single-end ChIP-seq data for H3K4me3 and H3K36me3 in the model organism Caenorhabditis elegans were downloaded from NCBI GEO with series IDs GSE28770 and GSE28776, respectively. These two histone modification marks have different distribution profiles, namely promoter-proximal enrichment with punctuated peaks for H3K4me3, and gene body enrichment with broad peaks for H3K36me323. The analysis was performed on a computer with Windows 7 Enterprise operating system (Microsoft Corporation, USA), 3.4 GHz CPU and 8 GB memory. Figure 1 shows the GUI displayed upon launching ChIPdig15 from R Studio. The left pane displays four radio buttons, each corresponding to one of the four analysis modules, as well a clickable box for selection of the folder containing the input files for the analysis.
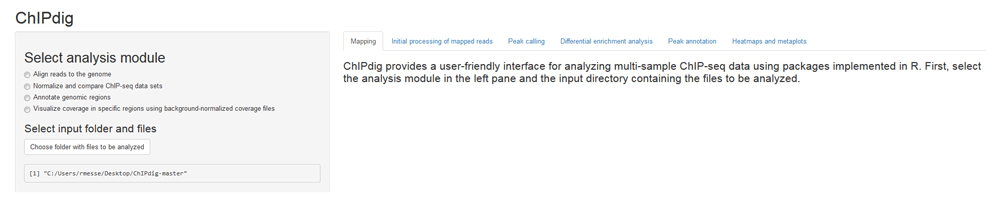
The left pane displays four radio buttons for selection of the analysis module and a clickable box for selection of the folder containing the input files. Additional features are sequentially unlocked as the user progresses through the software suite. Outputs generated throughout the analysis and additional instructions are placed in the main panel under a tab corresponding to the analysis module selected by the user.
The aligning module of ChIPdig relies primarily on the QuasR package, which supports the analysis of single-read and paired-end deep sequencing experiments17. If desired, read preprocessing can be performed to prepare the input sequence files prior to alignment, e.g. removal of sequence segments corresponding to adapters and low-quality reads. Sequence files in FASTQ format for each input DNA and ChIP sample corresponding to the H3K4me3 and H3K36me3 ChIP-seq experiments were aligned to the WS220/ce10 reference genome (available through the BSgenome package16) (Figure 2), producing an average of 10 million mapped reads per sample. The execution time was approximately 2 h 30 min.
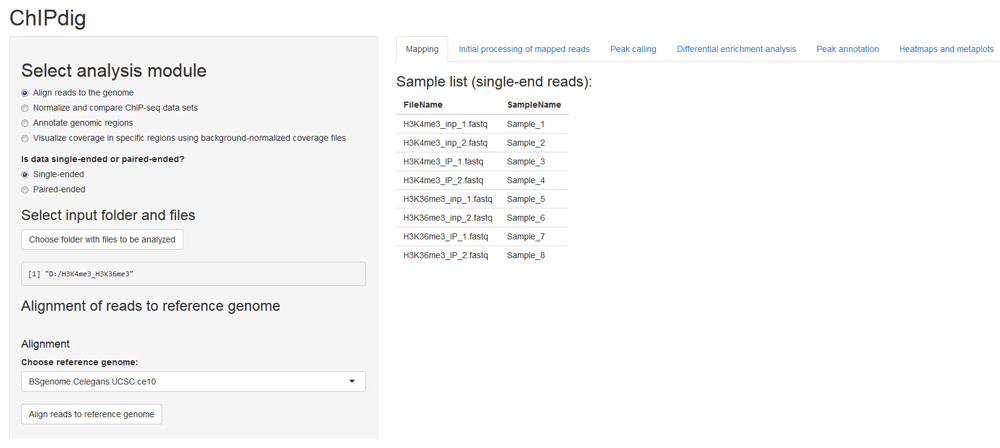
The user supplies a text file listing the input files corresponding to unmapped reads in either uncompressed (e.g.: '.fq', '.fastq') or compressed (e.g.: '.gz', '.bz2', '.xz') format. Both single-end and paired-end data are supported. A scroll-down menu displays the reference genome assemblies available for alignment.
A recurrent problem in ChIP-seq data analysis lies at the comparison of multiple coverage profiles generated from different experiments or corresponding to different conditions. This problem is addressed in the second analysis module of ChIPdig. Upon its selection, the user is prompted to provide a tab-delimited text file with the names of each ChIP sample (treatment) mapped reads file in BAM format, the matching input DNA (control) file, a sample ID, the condition or target name, and a color designation for the output peak and coverage files (Figure 3a). Any of the 657 R built-in colors can be chosen. A bin size parameter set to 50 bp (base pairs) by default is used in both peak calling and genome-wide library normalization for differential enrichment analysis. If desired, duplicate reads can be removed prior to normalization and sequences can be extended to a median fragment length that can be estimated either computationally or experimentally (e.g. average size of fragments generated by chromatin shearing) (Figure 3b). The initial mapped read processing outputs a table with library sizes and median fragment sizes, a chromosome size list, and a multidimensional scaling plot representing the similarity of samples in the data set (Figure 3c). Data are normalized based on library sizes and on the trimmed means of M values (TMM) approach24, which is implemented in the edgeR package13. Initial mapped read processing for the H3K4me3 and H3K36me3 ChIP-seq data took approximately 10 min.
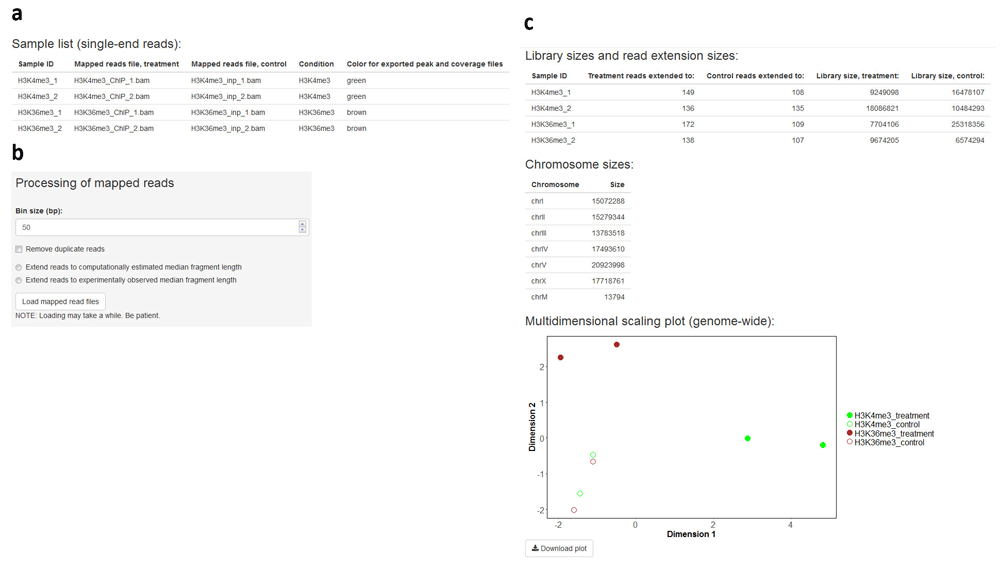
The user supplies a tab-delimited text file listing the input DNA and ChIP sample mapped read files (a). If desired, duplicate read removal and fragment size extension can be performed (b). The application outputs a table with library sizes and median fragment sizes, a chromosome size list, and a multidimensional scaling plot representing the similarity of samples in the data set (c).
Completion of the initial processing of mapped reads releases options for downstream processing which are posted to the sidebar panel, namely export of files representing sequencing coverage, peak calling and differential enrichment analysis (Figure 4). Coverage is expressed in log2-transformed counts per million (cpm) for each genomic bin and, for each sample ID indicated by the user, three files with bedGraph format are generated: treatment (ChIP sample), control (input DNA sample) and the treatment-to-control ratio. Each file can be loaded onto a genome browser for visualization (Figure 5). Generation of coverage files for H3K4me3 and H3K36me3 data took approximately 8 min.
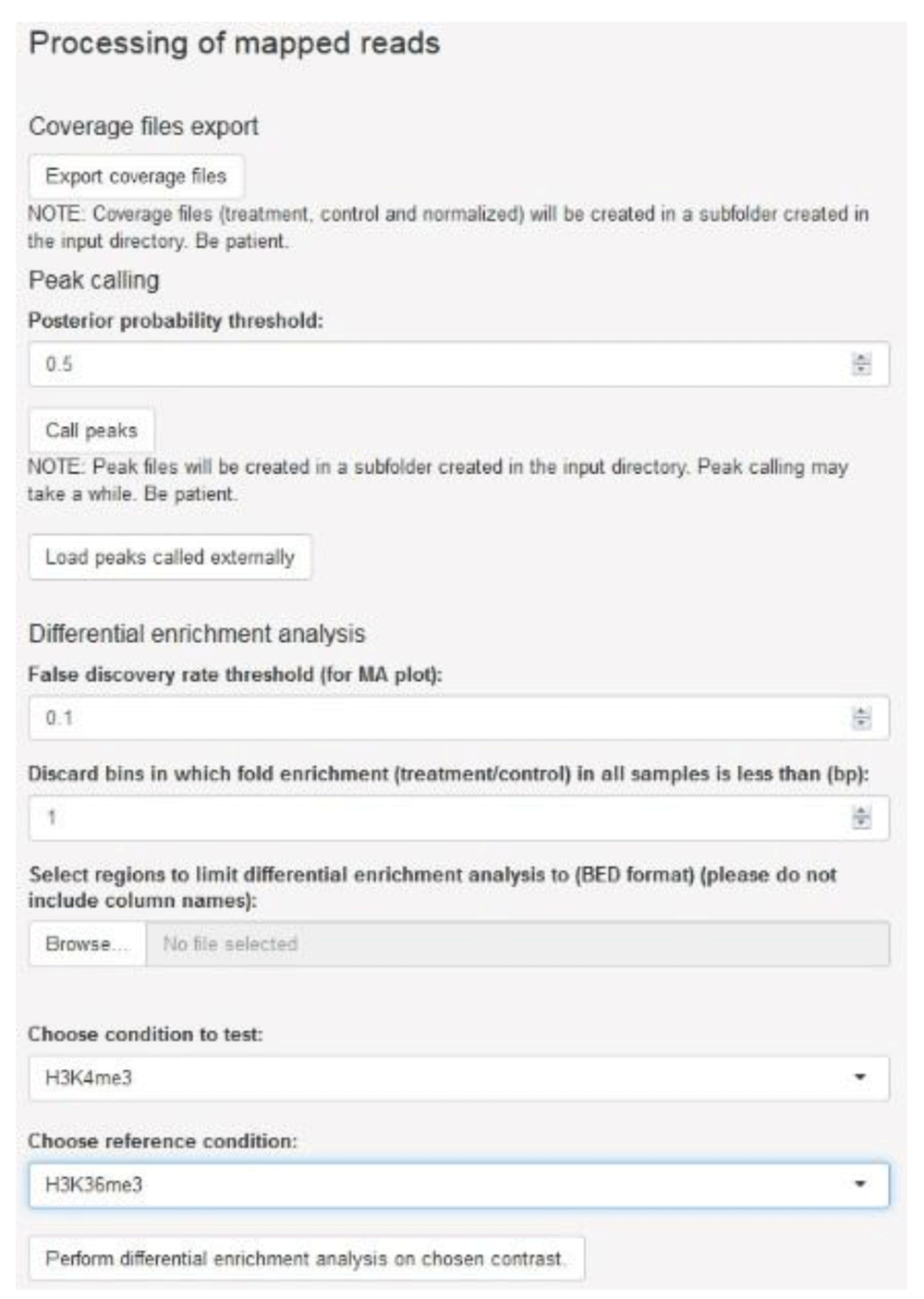
The user may generate coverage files in bedGraph format, perform peak calling for each sample, and assess differential enrichment for a set of genomic regions of interest.
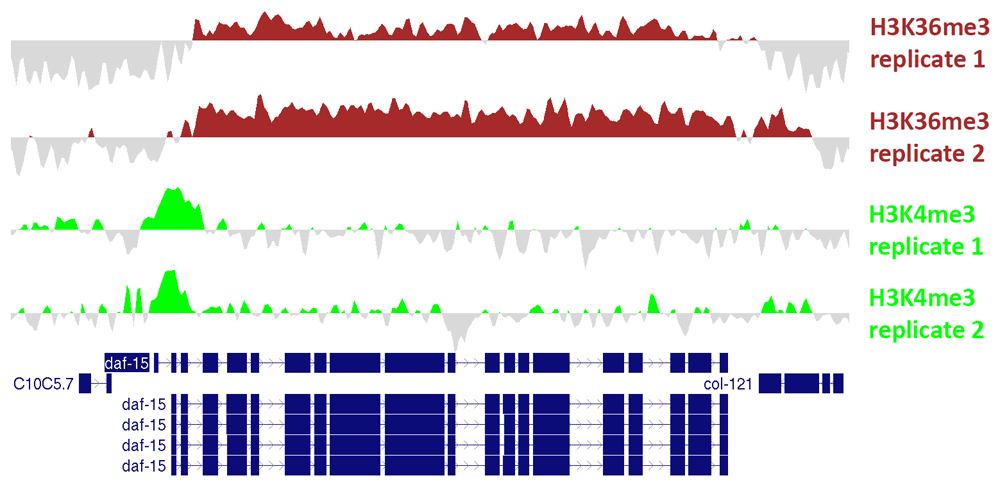
Values represent log2-transformed counts per million (cpm) for the ChIP sample subtracted by log2-transformed cpm for the input sample. Files in bedGraph format were exported from ChIPdig and loaded onto the UCSC Genome Browser. These profiles exemplify the well-known scenario whereby H3K4me3 accumulates around the transcription start site and H3K36me3 is enriched at the gene body.
Peak calling is performed via the BayesPeak package by using a hidden Markov model and Bayesian statistical methodology18. The user specifies a posterior probability (PP) threshold and genomic regions with PP above such threshold are identified as peaks. If replicates are indicated, commonly enriched genomic regions representing replicated peaks can be derived, and a track definition line can be added, allowing each file to be loaded onto the UCSC genome browser for visualization. In addition, a consensus peak set representing all candidate enriched regions across the full data set may be obtained. The user may be interested in choosing this option for posterior differential enrichment analysis. Peak calling was performed for the H3K4me3 and H3K36me3 ChIP-seq data sets, yielding 4994 replicated peaks for H3K4me3 and 25855 replicated peaks for H3K36me3. Peak calling execution time was approximately 12 h.
Differential enrichment analysis resorts to functions implemented in the csaw12 and edgeR13 packages. Following library size and TMM normalization, bin-level coverage is computed and, if desired, bins in which coverage for input DNA samples exceeds that of the corresponding ChIP samples are filtered off. If the user is interested in differential enrichment analysis in a specific set of regions, the corresponding file in BED format has to be provided. To illustrate this feature of ChIPdig, differential enrichment analysis was performed for comparing H3K4me3 coverage with that of H3K36me3 using either H3K4me3 replicated peak coordinates (Figure 6a) or those corresponding to H3K36me3 peaks (Figure 6b), with a false discovery rate (FDR) threshold of 0.1. As expected, at H3K4me3 genomic peak coordinates, H3K4me3 coverage is greater than that of H3K36me3, and the opposite is observed for H3K36me3 peaks. Differential enrichment analysis for both comparisons took approximately 5 min.
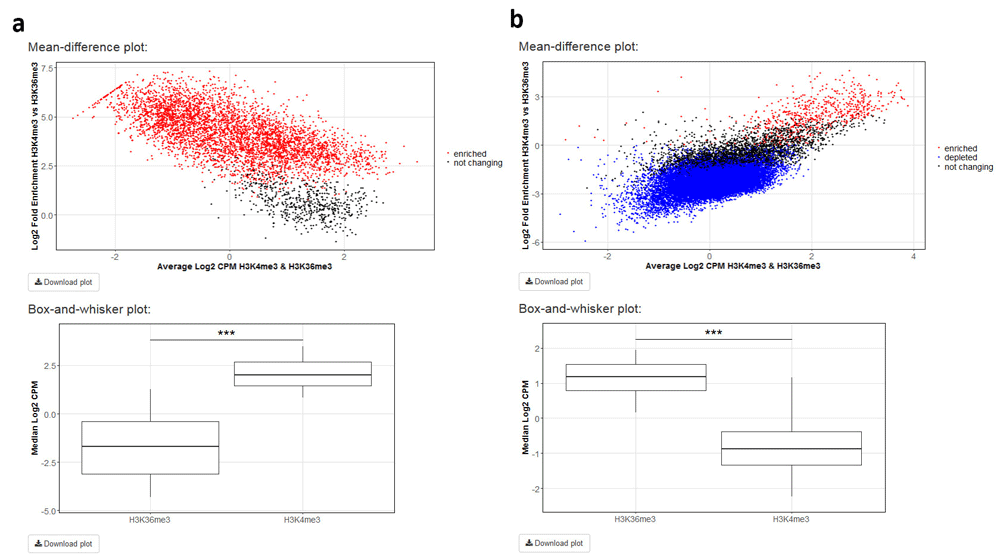
H3K4me3 ChIP-seq coverage was compared with that of H3K36me3 using either (a) H3K4me3 peak genomic coordinates or those of (b) H3K36me3 peaks with a false discovery rate threshold of 0.1. For each analysis, a mean-different plot representing the library size-adjusted log2-transformed fold change (the difference) against the average log2-transformed coverage (the mean), as well as a box-and-whisker plot showing the global change in normalized coverage between the two conditions, were originated.
Annotation of genomic regions of interest is performed via the ChIPseeker package19. The user supplies the file with regions in BED format and chooses the reference genome assembly, as well as the distance upstream and downstream of the annotated transcription start site to be considered for assignment of promoter regions (Figure 7a). Replicated H3K4me3 and H3K36me3 peaks were annotated and, characteristically of these marks23, most H3K4me3 peaks were assigned to promoters (Figure 7b), whereas H3K36me3 peaks lie predominantly at gene bodies (Figure 7c). The execution time for both annotation operations was approximately 3 min.
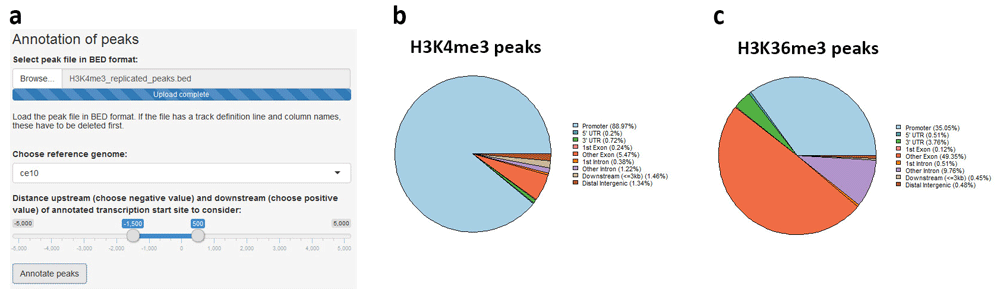
(a) The region file in BED format is uploaded and reference genome assembly, as well as distances upstream and downstream of transcription start site, are chosen. (b) Replicated H3K4me3 peaks are mostly promoter-proximal, whereas (c) H3K36me3 peaks are found predominantly at gene bodies.
The user can supply multiple coverage files in bedGraph format and generate heatmaps and metaplots to visualize coverage in a specific region set in a comparative manner. This module of ChIPdig relies on a custom algorithm which builds a coverage matrix based on bin size and reference coordinates (start, end or both) selected by the user. Such matrix is then plotted in the form of a comparative metaplot and a set of heatmaps. H3K4me3 and H3K36me3 coverage files were loaded onto ChIPdig, along with a BED file with C. elegans transcription units in the WS220/ce10 reference assembly. Coverage was computed in 25 bp bins and gene bodies were either expanded or compressed to 500 bp. Windows of 250 bp upstream and downstream of each region were selected (Figure 8a). Typical of H3K4me3, coverage is higher in the vicinity of the transcription start site, whereas H3K36me3 is enriched at transcription unit bodies (Figure 8b, c). Heatmap and metaplot generation took approximately 4 min.
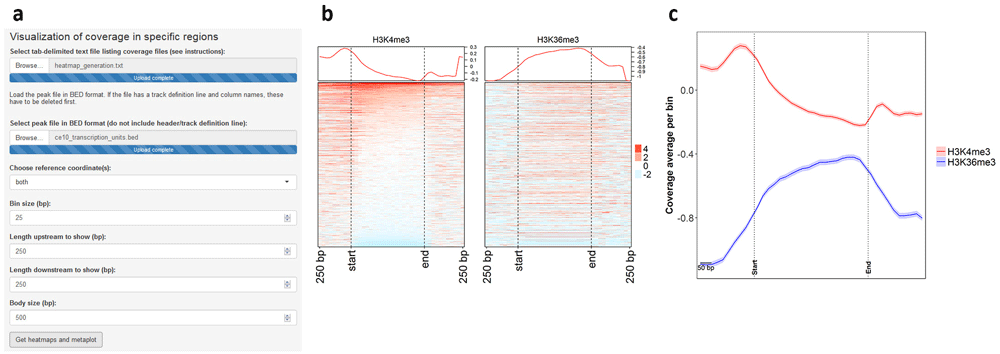
Coverage files for H3K4me3 and H3K36me3 were supplied to ChIPdig, as well as a file with coordinates of C. elegans transcription units. (a) The heatmaps and metaplot were generated by considering a 25-bp bin size, and gene bodies were resized to 500 bp. Windows of 250 bp upstream and downstream of each region were selected. As observed in both (b) the heatmaps and (c) the comparative metaplot, coverage for H3K4me3 is overall higher in the vicinity of transcription start sites, while that of H3K36me3 is enriched at transcription unit bodies.
In the past decade, several tools have been made available through Galaxy, an interactive system that provides a simple Web portal enabling users to analyze genomic data derived from high-throughput sequencing techniques25. We have compared results obtained by ChIPdig with those obtained within Galaxy using Bowtie3 for read alignment and MACS25,6 for peak calling. The percentage of mapped reads was higher for ChIPdig, but it should be noted that no read pre-processing was performed for read mapping in Galaxy (Figure 9a). For H3K4me3 peak calling, the number of called peaks using ChIPdig was similar to that in Galaxy, and, for H3K36me3, more peaks were called in ChIPdig than in Galaxy (Figure 9a). This can be attributed to the fact that the peak calling algorithm used by ChIPdig, and which is implemented in the BayesPeak package18, generates smaller peaks than those generated by Bowtie. More importantly, the spatial localization of ChIPdig-generated peaks overlapped very significantly with that of Galaxy-generated peaks (Figure 9b). We have also compared coverage tracks generated by ChIPdig (background-subtracted) with those generated by the Galaxy version of DeepTools and observed that the corresponding profiles are very similar (Figure 9c). These observations indicate that ChIP-seq data analysis using ChIPdig compares favorably with that in Galaxy.
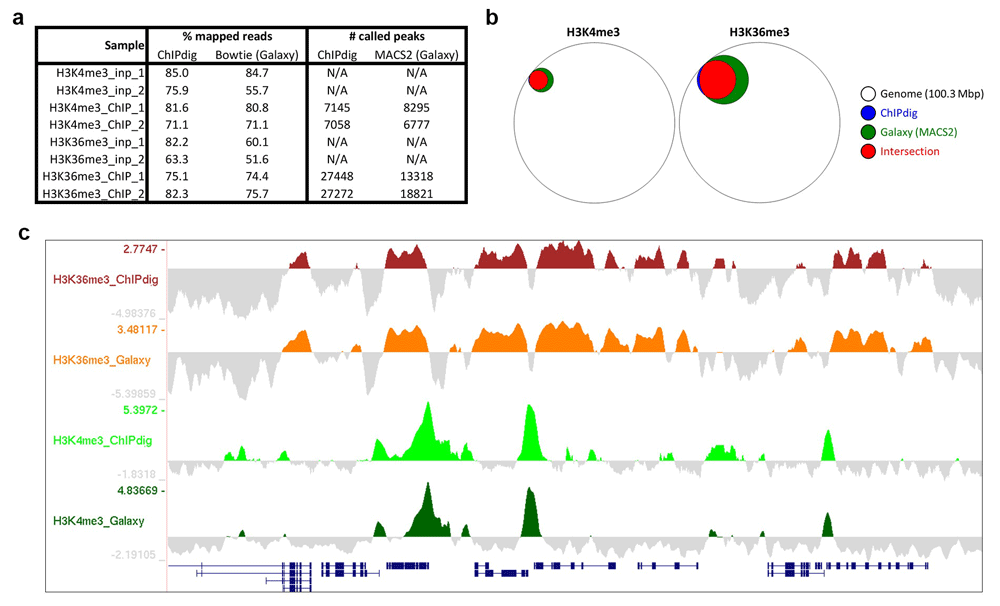
(a) The percentage of mapped reads using the alignment module of ChIPdig is higher than that using Bowtie within Galaxy, but it should be noted that read pre-processing was not included in the latter analysis. Peak calling in ChIPdig and in Galaxy resulted in similar numbers of peaks called for H3K4me3, and more H3K36me3 peaks were called using ChIPdig compared with Galaxy-generated peaks. (b) Importantly, ChIPdig-generated peaks overlapped very significantly with Galaxy-generated peaks. (c) Coverage tracks generated by ChIPdig have a profile similar to those generated by the Galaxy version of DeepTools.
ChIPdig15 is a user-friendly application for handling multiple ChIP-seq data sets and has diverse useful capabilities spanning a comprehensive analysis pipeline, namely: read alignment to a reference genome, ChIP-seq data normalization, peak calling, differential enrichment analysis, annotation of genomic regions, and generation of comparative heatmaps and metaplots for visualization of normalized coverage.
All data underlying the results are available as part of the article and no additional source data are required.
Source code available from: https://github.com/rmesse/ChIPdig.
Archived source code at time of publication: https://doi.org/10.5281/zenodo.334578815.
License: GNU General Public License 3.0.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)