Keywords
Histiocytic Disorders, Lumps/Bumps, malignant Neoplasms, benign Neoplasms, Skin signs of systemic disease
Histiocytic Disorders, Lumps/Bumps, malignant Neoplasms, benign Neoplasms, Skin signs of systemic disease
Cutaneous Langerhans cell histiocytosis (LCH) is a rare disorder manifest in the proliferation of cells with phenotypical characteristics of Langerhans cells which involves the cutaneous structures1. We have used the term ‘cutaneous’ in this review to differentiate from ‘skin-limited’ which implies the absence of systemic disease involvement1,2. The incidence of cutaneous LCH varies from two to nine cases per million children per year1,2. Rarely, the disease is present at birth or in the neonatal period. A proportion of these cases spontaneously resolve however no consistent variables have been identified which provide predictive value as to which cases will resolve or remain skin-limited, and which will manifest with multisystem LCH later in life1,2. The rate of progression of cutaneous LCH to other organs has varied widely in previous studies, from 0 to 60%1. This lack of accurate and reliable data makes it difficult to provide information to patients regarding the risk of progression of disease and limits the development of evidence-based screening measures to identify the presence of systemic disease. Currently, consensus guidelines1 state that most cases of cutaneous LCH spontaneously regress but some cases do progress to multisystem disease1. It is unclear whether cutaneous LCH is merely clinically more easily identified and hence often precedes diagnosis of internal disease. This would also suggest that widespread screening of cases of cutaneous LCH may produce lead-time bias in the survival rates of individuals with multisystem LCH with cutaneous involvement, an issue which to date has not been explored. Currently, in cases of cutaneous LCH screening is considered mandatory1,2.
Regarding identified risk factors for disease progression and mortality, overwhelmingly the data is sourced from cases of systemic LCH3,4, which may or may not include cutaneous disease. Data from older children also far exceeds data from neonatal cohorts, limiting or knowledge of differences between presentations in the neonatal population and older pediatric age groups. Only one retrospective case series of 19 patients5 examined survival outcomes in infants diagnosed with cutaneous LCH within the first 4 weeks of life, with long-term follow-up beyond 10 years being limited to small case series of less than 10 patients6. In the setting of systemic LCH, inadequate response to initial therapy and risk organ involvement, (defined as bone marrow, liver, spleen and/or lung), are the currently associated with adverse clinical outcomes and mortality in LCH1,2.
Isolated bone involvement portends significantly prolonged survival compared with other organ involvement3,4. As expected, patients with multiple organ involvement have been found to have the highest risk of progression and mortality7. Detection of the BRAF-V600E mutation (often seen in systemic LCH but rarely in skin-limited LCH), has also been associated with increased risk of disease recurrence4.
Overall, given the reports (albeit uncommon) of progression of cutaneous LCH to multisystem disease, the identification of clinical or histological predictive variables may reduce rates of unnecessary invasive screening in neonates with skin-limited LCH.
To collate all published cases of cutaneous congenital/neonatal LCH.
To perform a descriptive analysis of cases and reviews to evaluate mortality risk and risk of progression to systemic disease.
To identify risk factors which may contribute to mortality risk and risk of progression to systematic disease.
This systematic review was registered with PROSPERO (Registration number CRD42016041425) and was conducted in line with the PRISMA statement8
Information Sources for this review encompassed Medline (1946-March 1 2018), Embase (1980- March 1 2018) as well as “Epub ahead of print, and non-indexed citations” as shown in Figure 1. The search strategy is presented in Table 1. The databases searched were PubMed (National Library of Medicine), EMBASE, Cochrane Database of Systematic Reviews and published abstracts on Ovid Medicine (date limits for all: January 1 1980 to March 1 2018). The search terms used were: (Langerhans Cell Histiocytosis OR Hashimoto-Pritzker) AND (Congenital OR Birth OR Neonate) AND (Skin OR Cutaneous)
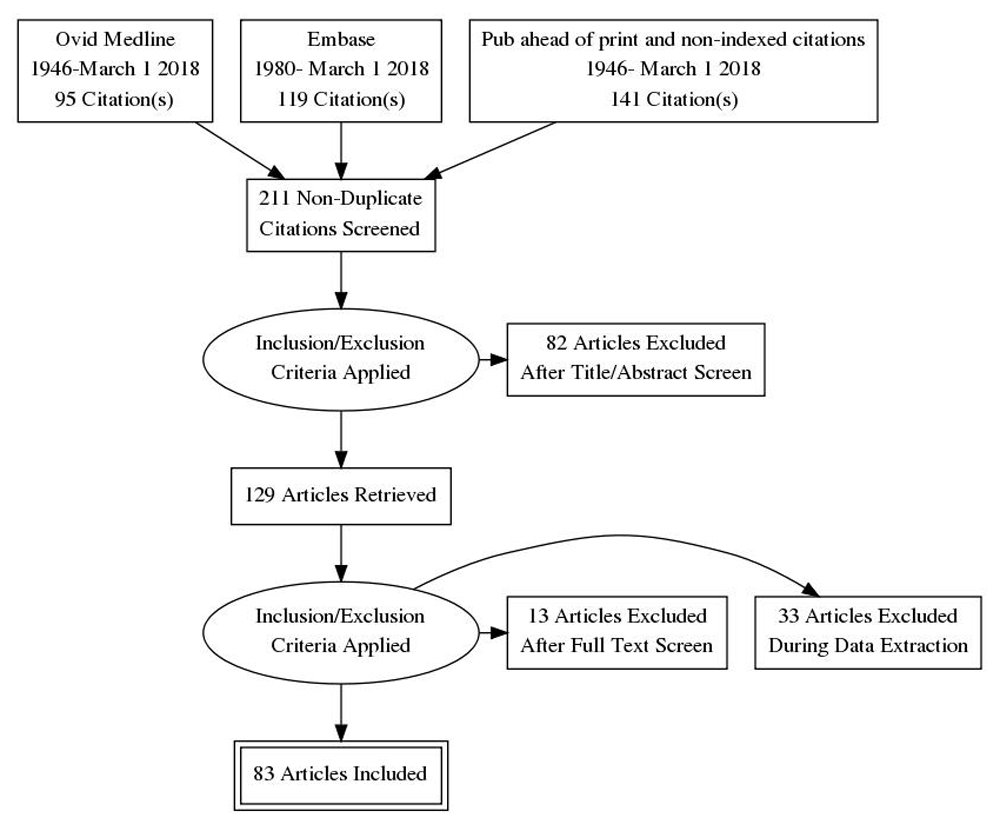
Eligibility criteria for this review included published case reports, case series and reviews with no restrictions of patient sex or ethnicity and language of publication. Eligible cases included:
1) Cases of histologically diagnosed LCH at birth (congenital) or within the first 4 weeks of life involving the skin.
2) Cases which report data pertaining to evidence of systemic involvement (clinical examination, skeletal survey etc.) and/or histological data (CD1a, eosinophil density etc.)
3) Cases with follow up data of any period.
Data collection was performed independently by two independent authors (EH and EY), with any disagreements regarding inclusion of citations being referred to a third author (VV) for mediation. Information was collected using a standardized data collection form (available as extended data on OSF9) with the principal outcomes of interest being mortality, age at demise and length of follow up. Data not available from the published article was requested via email contact with the relevant corresponding authors.
Potential sources of bias in collating cases were acknowledged including publications bias and reporting bias regarding the overall incidence of congenital and neonatal LCH, therefore only cases with a diagnosis of cutaneous LCH at birth (congenital) or within the first 4 weeks of life (neonatal) were included, and no attempt to quantify the number of cases of systemic LCH with a “missed” diagnosis of self-resolving cutaneous congenital LCH was undertaken. Particular effort was made to include unpublished cases and cases presented as posters and abstracts in order to reduce the impact of publication bias in our analyses.
An exploratory univariate analysis (using Pearson correlation coefficients for categorical variables and chi-squared tests for binary variables) was undertaken to correlate mortality and the progression to systemic disease with the clinical and histological variables collated.
A total of 211 articles were identified in the literature review; 82 of these articles were removed upon review of titles and abstracts against eligibility criteria. Full-text review of 129 articles excluded 12 review articles, 1 duplicated case report and 33 articles (containing 42 cases) due to lack of follow up data. The remaining 83 articles5,10–91 containing 128 individual cases were used as the basis of this review.
The summarized demographic data of the included cases is presented in Table 2.
The results of univariate correlation analysis are summarized in Table 3. The presence of multiple lesions was associated with an increased length of survival (r=0.304 p<0.05), whilst the presence of systemic disease portends a worse prognosis, with a statistically significant chi squared statistic (χ2 =14.96, 2DF p<0.001). Having lesions at birth had an odds ratio (OR) of mortality of 1.38, which did not reach statistical significance (95%CI=0.417-4.56). Individuals presenting with either weight loss, hepatosplenomegaly and internal organ involvement also had a worse prognosis and decreased overall survival (OR= 8.01 95% CI=2.07-30.86)
The presence of ulcerated lesions did not change risk of survival (OR=0.53 95% CI 0.11-2.05) Having less than 10 lesions increased the risk of mortality but not to a statistically significant degree (OR=1.77 95% CI= 0.76-17.30). Vesicular lesions were significantly more likely to be associated with mortality (OR=10.8 95% CI=2.83-41.26). Of 128 cases, 112 were screened for systemic involvement (87.5%). Of the screened cases, 66 were found to have cutaneous involvement only (51.6%). The mortality rate for those with identified systemic involvement was 27.4% (n= 17/62). The calculated OR for mortality based upon the presence of systemic involvement was 12.3 (95% CI). No statistically significant associations or OR were seen between histological markers and clinical outcomes including mortality or length of survival in the data examined. Given the heterogeneity of the sample, no multivariate analysis was performed on the collated data.
The results of this systematic review of case reports of cutaneous neonatal LCH differ from the pre- existing literature in several areas. This may be because existing data includes all cases of pediatric LCH, as opposed to the congenital and neonatal cases focused on in this review. This highlights the need for recognition that congenital and neonatal LCH have inherently different clinical characteristics compared to other pediatric cases of LCH. Minkov et al.3 have reported that the trunk was the most common overall site of disease. However, our data suggest that a large proportion of congenital and neonatal cases involve multiple anatomical sites (n=65). No significant gender predominance was identified (males=63; females=55). A weak association was seen between a later onset of disease and a worse prognosis (r=0.263, p<0.05). This is in line with the literature with earlier onset disease significantly associated with spontaneous resolution92,93.
Systemic disease was identified in 48.4% of cases (n=62) lower than the rates for the overall pediatric group at 59%3, and those reported by Stein et al. (63.1%)5. In line with previous research and recommendations5, systemic disease was significantly correlated with mortality (r=0.453, p<0.05), with persistent cutaneous lesions associated with poorer outcomes3,75,92. The overall mortality rate for all cases in the population of this review was 14.05%.
Previous studies have suggested high rates of spontaneous clinical remission (from 60%3 to 100%5) in skin-limited LCH, and 8% in multisystem disease94. The accuracy of such figures is disputed due to the absence of systemic screening and long term follow up in these published reports. We attempted to identify cases of spontaneous remission (both clinical and biological) in the literature. Clinical remission was documented in 41/128 cases (32.1%); however, due to the high variability in length of follow-up and low rates of systemic screening post clinical remission, rates of biological remission could not be accurately established. We would suggest that long term prospective follow-up studies with systemic screening (both at diagnosis and post clinical remission) are required to accurately quantify rates of spontaneous biological remission in future studies.
Regarding lesion morphology, Battistella et al.92 suggest that single, necrotic, hypopigmented macules and distal topography (lesions present at a distal site) suggest a self-regressive form of disease92. This is still an area of contention with no reliable data from cohorts larger than 20 patients5,93,95–98. We identified a weak correlation between skin lesion descriptors and overall mortality as well as length of survival in the neonatal and congenital LCH population. The presence of multiple lesions was associated with increased length of survival, although the presence of lead-time bias was likely given the non-significant differences in mortality between the two groups. Vesicular lesions were associated with increased mortality whereas no impact of survival was seen in the presence of ulcerated lesions. We anticipated that reporting bias would result in confirmation of an association between ulcerated lesions and mortality if one existed, although this has not been confirmed by our review. One explanation is that vesicular lesions commonly progress to ulceration during the stages of healing, thus emphasizing the need for consistent descriptors in case reports of LCH. Alternatively, ulcerated lesions might have an association with mortality but not in the congenital and neonatal LCH cohort.
Most cases identified were congenital (67.9%; n=87), although some controversy exists regarding whether congenital cases exist at all93. Morren states that LCH presents prior to 3 months of age but does not occur congenitally93. Given the retrospective nature of our study, we were unable to shed further light on this debate as we were reliant upon multiple authors’ observations and recordings.
Given the variability in patient follow-up in this review, the current estimates of mortality risk are only valid until 18 months of age (the mean length of follow-up). The lack of long-term follow-up is the major reason why data is lacking regarding long term recurrence rates in neonatal LCH and thus our review is limited to conclusions regarding short- and medium-term outcomes.
Future research should expand upon this by analyzing longer-term outcomes. Haupt1 has recommended a long-term follow-up of 5 years for patients, mirroring that of childhood cancer survivors. This is applicable even to both skin-limited LCH and systemic disease. Progression of skin-limited LCH to multisystem involvement is documented in the literature1,5.
We had a limited ability to identify statistically significant variables that contribute to LCH mortality due to limited follow-up in documented cases. The GRADE approach99 to assessing the quality of evidence and strength of recommendations (available as extended data on OSF9) shows the absence of control groups, and incomplete follow up. Long-term, prospective, multicenter collaborative studies needed to confirm the findings of this review and are important steps in characterizing the progression of neonatal LCH.
We present a systematic review of case reports of cutaneous congenital and neonatal LCH. The descriptive characteristics in this review significantly differ from descriptions of overall pediatric LCH, highlighting the clinical differences between these entities. Congenital and neonatal LCH most commonly presents in multiple anatomical sites at or shortly after birth, with the presence or absence of systemic involvement significantly impacting mortality. Further prospective, long-term multicenter collaborative studies are required to corroborate the results of this review.
The Data Collection Proforma and GRADE Bias Assessment are available on OSF. DOI: https://doi.org/10.17605/OSF.IO/TRX429.
Data are available under the terms of the Creative Commons Zero "No rights reserved" data waiver (CC0 1.0 Public domain dedication).
A completed PRISMA Checklist for this study is available on OSF. DOI: https://doi.org/10.17605/OSF.IO/TRX429.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)