Keywords
benchmarking, high-dimensional cytometry, Bioconductor, ExperimentHub, clustering, differential analyses
This article is included in the Bioconductor gateway.
This article is included in the RPackage gateway.
benchmarking, high-dimensional cytometry, Bioconductor, ExperimentHub, clustering, differential analyses
We have revised the manuscript, package vignettes, and package help files to address the issues raised by the reviewers. In particular, we have added two new vignettes titled (i) “Examples and use cases”, which includes reproducible code for the example previously included in the text, as well as new examples on clustering and differential analyses, and (ii) “Contribution guidelines”, which explains the procedure and required data files for contributing new datasets. The text has been clarified in a number of locations to better explain the motivation for creating the HDCytoData package, and more clearly explain aspects that may be non-intuitive for users who are less familiar with high-dimensional flow and mass cytometry data. Specific responses to the issues raised by the reviewers are listed in the responses to the reviewers.
See the authors' detailed response to the review by Laurent Gatto
See the authors' detailed response to the review by Shila Ghazanfar
Benchmarking analyses are frequently used to evaluate and compare the performance of computational methods, for example by users interested in selecting a suitable method, or by developers to demonstrate performance improvements of a newly developed method. A critical part of any benchmark is the selection of appropriate benchmark datasets1,2. In some cases, suitable publicly available datasets may be found in the literature. Alternatively, new experimental or simulated datasets containing a known ground truth may be created by the authors of the benchmark1,2.
High-dimensional cytometry refers to a set of recently developed technologies that enable measurement of expression levels of up to dozens of proteins in hundreds to thousands of cells per second, using targeted antibodies labeled with various types of reporter tags. This includes multi-color flow cytometry, mass cytometry (or CyTOF), and sequence-based cytometry (or genomic cytometry). Due to the large size and high dimensionality of the resulting data, numerous computational methods have been developed for analyzing these datasets3. Many of these methods are based on the fundamental concept of analyzing cells in terms of cell populations, for example using clustering to define cell populations, or detecting differential cell populations between conditions.
In our previous work, we have collected a number of benchmark datasets to evaluate methods for clustering4 and differential analyses5 in high-dimensional cytometry data. This includes publicly available datasets previously published by other groups or our experimental collaborators, as well as new semi-simulated datasets that we generated. In these previous publications, we recorded links to original data sources and made all data available via FlowRepository6. FlowRepository is a widely used resource in the cytometry community, which provides a permanent record of publicly available datasets associated with peer-reviewed publications, and which has also been used by other authors to distribute benchmark datasets (e.g., 7,8). However, FlowRepository is primarily accessed via a web interface, and downloading and loading data for further analysis in R requires customized code and matching of metadata (e.g., sample information), which can hinder accessibility and reproducibility.
Here, we introduce the HDCytoData package, which provides a resource for re-distributing high-dimensional cytometry benchmark datasets through Bioconductor’s ExperimentHub9, in order to improve accessibility. ExperimentHub provides a flexible platform for hosting datasets in the form of R/Bioconductor objects, which can be directly loaded within an R session. We have formatted the datasets in HDCytoData into standard SummarizedExperiment and flowSet Bioconductor object formats10–12, which include all required metadata within the objects and facilitate interoperability with R/Bioconductor-based workflows. The data objects are intended to be static, with no major updates following release. We envisage that these datasets will be useful for future benchmarking studies, as well as other activities such as teaching, examples, and tutorials. The package is extensible, allowing new datasets to be contributed by ourselves or other researchers in the future. It is designed to be accessible for users who are familiar with R and Bioconductor, but who may not have used ExperimentHub packages before. The package is freely available from http://bioconductor.org/packages/HDCytoData.
The benchmark datasets currently included in the HDCytoData package consist of experimental and semi-simulated data, and can be grouped into datasets useful for benchmarking algorithms for (i) clustering and (ii) differential analyses. Table 1 and Table 2 provide an overview of the datasets.
For more details on these datasets, see Table 2 in 4, or the HDCytoData help files.
| Dataset | ExperimentHub ID | Number of cells | Number of dimensions | Number of reference cell populations | Type of ground truth | FlowRepository ID | Original reference |
|---|---|---|---|---|---|---|---|
| Levine_ 32dim | EH2240 – EH2241 | 265,627 | 32 | 14 | Manual gating | FR-FCM-ZZPH | 13 |
| Levine_ 13dim | EH2242 – EH2243 | 167,044 | 13 | 24 | Manual gating | FR-FCM-ZZPH | 13 |
| Samusik_ 01 | EH2244 – EH2245 | 86,864 | 39 | 24 | Manual gating | FR-FCM-ZZPH | 14 |
| Samusik_ all | EH2246 – EH2247 | 841,644 | 39 | 24 | Manual gating | FR-FCM-ZZPH | 14 |
| Nilsson_ rare | EH2248 – EH2249 | 44,140 | 13 | 1 (rare population) | Manual gating | FR-FCM-ZZPH | 15 |
| Mosmann_ rare | EH2250 – EH2251 | 396,460 | 14 | 1 (rare population) | Manual gating | FR-FCM-ZZPH | 16 |
For more details on these datasets, see Supplementary Note 1 in 5, or the HDCytoData help files.
| Dataset | ExperimentHub ID | Type of data | Number of cells | Number of dimensions | Type of ground truth | Type of differential analysis | FlowRepository ID | Original reference |
|---|---|---|---|---|---|---|---|---|
| Krieg_Anti_ PD_1 | EH2252 – EH2253 | Experimental | 85,715 | 24 (cell type) | Qualitative | Differential abundance | FR-FCM-ZYL8 | 17 |
| Bodenmiller_ BCR_XL | EH2254 – EH2255 | Experimental | 172,791 | 24 (10 cell type; 14 cell state) | Qualitative | Differential states | FR-FCM-ZYL8 | 18 |
| Weber_AML_ sim | EH3025 – EH3046 | Semi- simulated (multiple simulation scenarios) | 157,593 (excluding spike-in) | 16 (cell type) | Spike-in cell labels | Differential abundance | FR-FCM-ZYL8 | 5 |
| Weber_BCR_ XL_sim | EH3047 – EH3064 | Semi- simulated (multiple simulation scenarios) | 85,331 (main simulation; excluding spike-in) | 24 (10 cell type; 14 cell state) | Spike-in cell labels | Differential states | FR-FCM-ZYL8 | 5 |
The raw datasets were collected from various sources (Table 1 and Table 2), and have been extensively reformatted and documented for inclusion in the HDCytoData package. Each dataset is stored in both SummarizedExperiment and flowSet formats, since these are the most commonly used R/Bioconductor data structures for high-dimensional cytometry data (and there is generally no straightforward way to convert between the two). The objects each contain one or more tables of expression values, as well as all required metadata. Following standard conventions used for cytometry data19, rows contain cells, and columns contain protein markers. Row metadata includes sample IDs, group IDs, patient IDs, reference cell population labels (where available), and labels identifying ‘spiked in’ cells (where available). Column metadata includes channel names, protein marker names, and protein marker classes (cell type, cell state, as well as non protein marker columns). Note that raw expression values should be transformed prior to performing any downstream analyses. Standard transformations include the inverse hyperbolic sine (asinh) with cofactor parameter equal to 5 for mass cytometry or 150 for flow cytometry data (20, Supplementary Figure S2); several other alternatives also exist21.
Most of these datasets include a known ground truth, enabling the calculation of statistical performance metrics. The ground truth information consists of reference cell population labels for the clustering datasets, and labels identifying computationally ‘spiked in’ cells for the differential analysis datasets. The datasets without a ground truth instead consist of experimental datasets that contain a known biological signal, which can be used to evaluate methods in qualitative terms; i.e., whether methods can reproduce the known biological result.
Extensive documentation is available via the help files for each dataset—including descriptions of the datasets, details on accessor functions required to access the expression tables and metadata, and links to original sources. In addition, reproducible R scripts demonstrating how the formatted SummarizedExperiment and flowSet objects were generated from the original raw data files from FlowRepository are included within the source code of the package.
New datasets may be contributed by ourselves or other authors in the future. The procedure for external contributions is described in the vignette titled “Contribution guidelines”, available from Bioconductor. This vignette describes the submission procedure (via GitHub), as well as the required files (data objects in SummarizedExperiment and flowSet formats containing all necessary metadata, reproducible R scripts showing how the formatted objects were generated from the original raw data files, documentation, and package metadata).
The HDCytoData package can be installed by following standard Bioconductor package installation procedures. All datasets listed in Table 1 and Table 2 are available in Bioconductor version 3.10 and above. Minimum system requirements include a recent version of R (3.6 or later; this paper was prepared using R version 3.6.1), on a Mac, Windows, or Linux system. Example installation code is shown below.
# install BiocManager install.packages("BiocManager") # install HDCytoData package BiocManager::install("HDCytoData")
Once the HDCytoData package is installed, the datasets can be downloaded from ExperimentHub and loaded directly into an R session using only a few lines of R code. This can be done by either (i) referring to named functions for each dataset, or (ii) creating an ExperimentHub instance and referring to the dataset IDs. Example code for each option for one of the datasets is shown below. Note that each dataset is available in both SummarizedExperiment and flowSet formats. After an object has been downloaded, the ExperimentHub client stores it in a local cache for faster retrieval. File sizes for these datasets range from 2.4 MB (Nilsson_rare) to 194.5 MB (Samusik_all) (see help files). The local download cache can be cleared using the removeCache function from the ExperimentHub package (see HDCytoData package help file or main vignette). For more details on accessing ExperimentHub resources, refer to the ExperimentHub vignette available from Bioconductor.
# load HDCytoData package library(HDCytoData) # option 1: load datasets using named functions d_SE <- Bodenmiller_BCR_XL_SE() d_flowSet <- Bodenmiller_BCR_XL_flowSet() # option 2: load datasets by creating ExperimentHub instance ehub <- ExperimentHub() query(ehub, "HDCytoData") d_SE <- ehub[["EH2254"]] d_flowSet <- ehub[["EH2255"]]
Once the datasets have been downloaded and loaded, they are available to the user as R objects within the R session. They can then be inspected and manipulated using standard accessor and subsetting functions (for either the SummarizedExperiment or flowSet object class). Example code to inspect a SummarizedExperiment is displayed below. For more details on how to load and inspect datasets, including the expected output from each function shown here, refer to the HDCytoData package main vignette available from Bioconductor.
# inspect SummarizedExperiment object d_SE assays(d_SE) rowData(d_SE) colData(d_SE) metadata(d_SE)
Documentation describing each dataset is available in the help files for the objects, which can be accessed using the standard R help interface, as shown below.
# display documentation (help files) ?Bodenmiller_BCR_XL help(Bodenmiller_BCR_XL)
The datasets currently included in the HDCytoData package (Table 1 and Table 2) can be used to benchmark methods for either (i) clustering or (ii) differential analyses. In addition, these datasets may be useful for other activities such as teaching, examples, and tutorials (e.g., demonstrating how to use a new computational tool).
For the clustering benchmark datasets (Table 1), performance can be evaluated by calculating metrics such as the mean F1 score or adjusted Rand index, which measure the similarity between two sets of cell labels (i.e., the cluster labels and the ground truth or reference cell population labels)1. A short example is shown in the vignette titled “Examples and use cases”, available from Bioconductor. For more extensive examples and evaluations, see the GitHub repository accompanying our previous study4.
These datasets can also be used to generate visualizations demonstrating the performance of dimension reduction algorithms. For example, Figure 1 compares three different dimension reduction algorithms (principal component analysis [PCA], t-distributed stochastic neighbor embedding [tSNE]22,23, and uniform manifold approximation and projection [UMAP]24,25), for one of the datasets (Levine_32dim), with colors indicating the ground truth cell population labels. The figure shows a clear visual separation between the populations, with varying performance for the different algorithms. Reproducible R code for this figure is available in the “Examples and use cases” vignette, and the GitHub repository http://github.com/lmweber/HDCytoData-example.
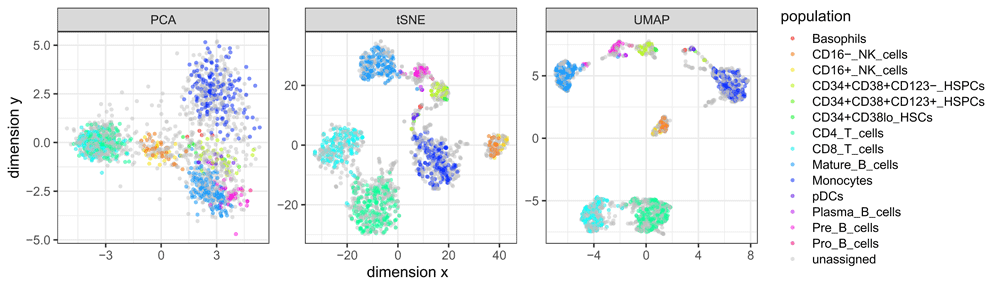
This example compares three different dimension reduction algorithms — principal component analysis (PCA), t-distributed stochastic neighbor embedding (tSNE), and uniform manifold approximation and projection (UMAP) — for visualizing cell populations in the Levine_32dim dataset (Table 1). Colors indicate the known ground truth cell populations.
For the differential analysis benchmark datasets (Table 2), methods can be evaluated by their ability to recover the known differential signals, either in quantitative terms using the ground truth spike-in cell labels (for the semi-simulated datasets), or in qualitative terms (for the experimental datasets). The differential signals consist of either differential abundance of cell populations, or differential states within cell populations (i.e., differential expression of additional functional markers within cell populations), providing conceptually distinct differential analysis tasks. A short example showing how to perform differential analyses on these datasets is provided in the “Examples and use cases” vignette. For more extensive examples and evaluations, see the GitHub repository accompanying our previous study5.
The HDCytoData package is an extensible resource providing streamlined access to a number of publicly available benchmark datasets used in our previous work on high-dimensional cytometry data analysis. Datasets are provided in standard Bioconductor object formats, and are hosted on Bioconductor’s ExperimentHub platform. In the future, it may make sense to develop similar packages for other data types, e.g., imaging mass cytometry, once several well-characterized benchmark datasets become available. By facilitating access to these datasets, we hope they will be useful for other researchers interested in designing rigorous benchmarks for method development or other computational analyses, as well as other activities such as teaching, examples, and tutorials.
All data underlying the results are available as part of the article and no additional source data are required.
Software available from: http://bioconductor.org/packages/HDCytoData
Source code available from: https://github.com/lmweber/HDCytoData
Archived source code at time of publication: https://doi.org/10.5281/zenodo.355105126
Licence: MIT License
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)