Keywords
SNP, Disease, Python, Bioinformatics
This article is included in the Python collection.
SNP, Disease, Python, Bioinformatics
In recent years, the number of cases of genetically originated diseases has increased, alarming the world and sparking interest in the development of precision medicine using molecular biomarkers. Single nucleotide polymorphisms (SNPs), the most common genetic difference among individuals, occurs in the human genome. These randomized modifications in DNA bases cause alterations in protein sequence residues of amino acids, thus altering their functions, which leads to different disease conditions in individuals1. Several of these SNPs have been identified as disease-related genetic markers that have been used to recognize genes responsible for particular diseases in humans2.
Distinguishing the evidence and the interpretation of a rich range of markers will be necessary to relate major alterations in the SNPs and to discover their connection in the progression of disease. Clarification of the phenotypic-associative mechanisms for these variations is therefore vital for comprehending the sub-atomic subtleties of disease origin and for developing novel therapeutic methods3,4.
Although SNPs may exist in various areas of the gene, such as promoters, introns, 5′-and 3′ UTRs, to date, most research has focused on disease-associated SNPs in coding regions or exons, especially non-synonymous SNPs, which may alter the biochemical ability of encoded proteins. In turn, altering gene promoters impact gene expression by changing transcription, binding transcription factor, methylation of DNA and modifications of histones. As a consequence, changes in gene expression, their impact on disease susceptibility, and drug responses can differ depending on the location of the SNP5–7.
With the expansion of genetic variants, different software could be used to generate new knowledge to support disease diagnosis and drug response studies and to develop new biomarkers for disease identification and drug customization. In this regard, a number of software applications have been developed in the last few years to classify, prioritize and evaluate the impact of genomic variants. For example, the Ensembl Variant Effect Predictor offers access to a large range of genomic annotations, with a variety of frameworks that answer different needs, with easy setup and evaluation methods8. Similarly, SnpEff categorizes the results of genome sequence variations, annotates variants according to their genomic location and estimates the coding effects. Depending on genome annotation, it is possible to predict coding effects such as non-synonymous or synonymous substitution of amino acids, stop codon gains or losses, start codon gains or losses, or frame changes9.
On the other hand, another tool, PolyPhen-2, assesses the potential impact of the genetic substitution of amino acids on the basis of physical, evolutionary comparative factors and model structural changes. Based on these profiles, the probability of a missense mutation becoming dangerous is measured on the basis of a combination of all these properties10. Similarly, SIFT calculates whether the substitution of amino acids affects protein activity, based on the homology of sequences and the physical properties of amino acids. It may be used for non-synonymous polymorphisms and laboratory-induced missense mutations that naturally occur, to effectively classify the effects of SNPs as well as other types, including multiple nucleotide polymorphisms11.
Moreover, Phyre2 is a web-based suite of tools for predicting and analysing protein structure, function and mutations. It has sophisticated remote homology identification methods to build 3D models, anticipate ligand binding sites, and evaluate the effect of amino acid variants, e.g. non-synonymous SNPs12. Missense 3D uses the user-provided UniProt ID of the query protein, wild-type residue and substitution and other information to generate PDB residue mapping and predict the substitution effect on the 3D protein structure13.
To conclude the effect and possible phenotype of SNP, these software and web applications require minimum information such as SNP genomic position, SNP ID, allele form, and/or gene name. Acquiring this information requires using different computational tools, extensive time and some analysis skills. Most of the time, only gene sequences are available in which the SNPs are hidden without any additional information.
We therefore introduce SNPector, a standalone SNP inspection software that can be used to diagnose gene pathogenicity and drug reaction in naked genomic sequences. SNPector identifies and extracts gene-related SNPs, and reports their genomic position, associated phenotype disorder, associated diseases, linkage disequilibrium, in addition to various drug reaction information. It detects and verifies the existence of an SNP in a given DNA sequence based on different clinically relevant SNP databases, such as NCBI ClinVar14, AWESOME15, and PharmGKB16. Lastly, it connects identified SNPs, related diseases and drugs, and produces numerous visualization figures to explain these relationships with the support of different Python modules.
The SNPector Python tool uses many packages to inspect the existence of SNPs in a given sequence. Moreover, SNPector provides users with detailed visualization figures, highlighting other SNPs with similar mutation effects on protein phosphorylation, ubiquitination, methylation, or sumoylation sites, and predicts substrates of N-acetyltransferase.
Additionally, SNPector provides the ability to visualize obtained information about the linkage disequilibrium of detected SNPs using various Python packages, such as Matplotlib17, generating a number of figures that summarize vast amounts of previously published data indicating SNPs allelic segregation, association, minor allele frequency. Figure 3 shows an example of illustrations that can be generated through SNPector.
SNPector was written using Pythpn3 programming language as a standalone package and can be run on different operating systems platforms supported with Python 3.x compilers. To achieve user-friendly usage, the SNPector only accepts input from FASTA sequence (Figure 1) and can be operated from a console through simple command line (Figure 2).

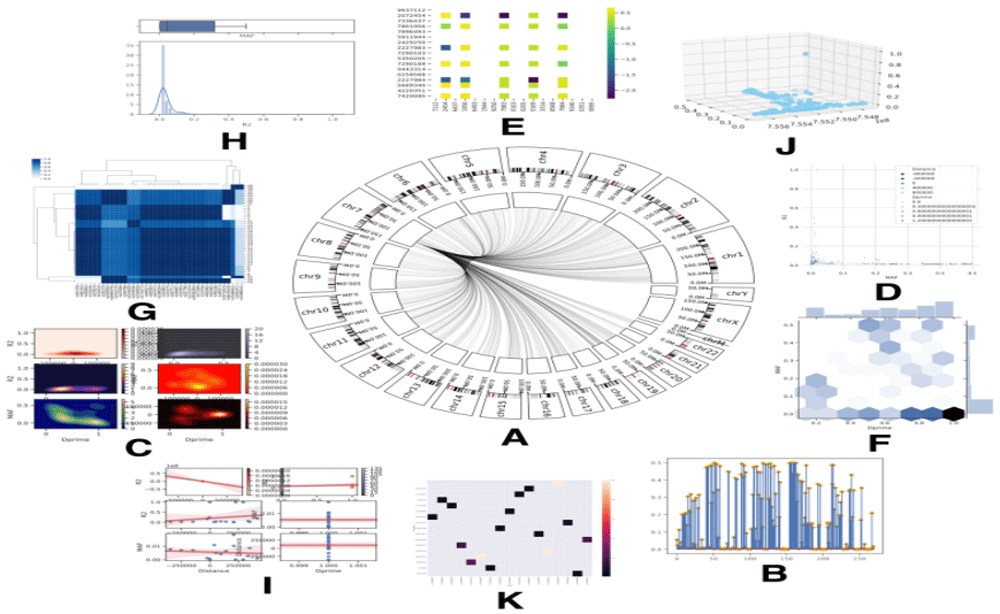
(A) Circos illustration where other SNPs that have same proprieties are located. (B) Lollipop plot shows values by vertical columns (C) Counter Plot between two values creating a different coloured shade in which more contrast means higher value. (D) Numerical schematic showing the distribution between four values by plotting and scaling colour contrast according to other to values. (E) Heat map between SNP linkage disequilibrium matrix to show how two SNPs are linked. (F) Marginal plot combining column graph and plot, both showing the relationship between two values. (G) Dendrogram with heat-map showing how all SNP are linked to each other. (H) Histogram with box plot to compare visually between two values. (I) Plot illustrating the regression fit of two plotted value. (J) 3D plot of three values. (K) Annotated heat-map showing the plotted value.
SNPector uses different SNP record information collected from NCBI ClinVar (159,184 records), AWESOME (1,080,551 records), and PharmGKB18 (3,932 records). Ldlink is an online tool that can be used to assess linkage imbalance (LD) throughout ancestral populations and is a popular method of exploring population-specific genetic framework and functionally navigation disease susceptibility areas19. In SNPector, an Application Program Interface (API) has been programmed to download an LDhap file containing linkage disequilibrium statistics and potentially functional variants for a query variant resulting from the inputted FASTA sequence.
SNPector starts by running BLAST20 software locally to find out the genomic location of a given DNA sequence on human genome. If successful, it retrieves the SNP records located within the query genomic range using NCBI ClinVar. According to retrieved records from the database, the detected SNPs in user-provided queries are marked as wild or mutated. Additionally, more information regarding detecting SNPs records will be retrieved from different implemented databases. This information will be used to generate different illustration figures.
If the process is successfully finished, SNPector will generate four different files: (A) Text file containing the output BLAST result, where the genomic location of the user-defined sequences is predicted; (B) tab delimited file containing SNPs retrieved by NCBI ClinVar located in the same regions; (C) two files regarding specific SNPs information retrieved from AWESOME and PharmGKB databases; (D) different figures depicting SNPs with a similar mutation effect to the detected SNPs located on other genomic regions, SNP linkage disequilibrium, the relationship between SNP, drug, and phenotype (Figure 3).
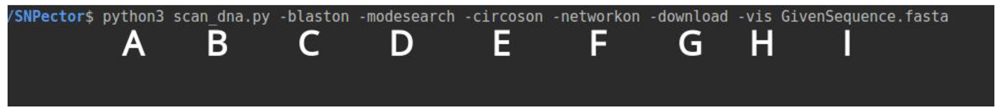
To achive maximum user-freindly usage, SNPector can be run and controlled by command line. SNPector command line structure (Figure 2) is as follows: A) Python3 compiler; B) scan_dna.py: program main script that contain all functions; C) -blaston / -blastoff: in order to initiate BLAST process to provide sequence alignment against the genome to locate where the sequence is situated, if the -blastoff is chosen it will use previous BLAST results; D) -modescan: to scan the given sequence and find out whether SNP occurs or exists in sequence or not, and -modesearch: to extract all SNPs occur in this range of sequence regardless they are exist or not; E) -circoson: draws a Circos figure to illustrate where SNPs with same properties/effect are located; F) -networkon: in order to link between SNPs, diseases and drugs and produces network HTML file; G) -download: activates the API to download data for identified SNPs from LDlink database; H) -vis: produces different figures and plots; I) GivenSequence.fasta: Tte user-provided sequence in FASTA file format. Any of the previous parameters can be deactivated when replaced with -off.
In this section we provide an example on how to use SNPector to extract SNPs from a naked given sequence without a reference sequence and how these extracted SNPs are linked to disease development and how they affect drug response. We show how to define the arguments of the SNPector function, interpret the results, and make visualizations.
We use part of an EGFR gene sequence downloaded from NCBI nucleotide database in FASTA format as shown in Extended data: File 121. The EGFR gene FASTA sequence submitted to NCBI contains SNPs that have a clinical effect involved in disease development, such as breast cancer. SNPector uses: (i) NCBI ClinVar database that describes SNP chromosome, position, ID, reference nucleotide, alternative nucleotide, quality, filter, and information to compare and detect SNPs in EGFR sequence that has clinical complications; (ii) PharmGKB database to investigate the SNP effect on disease development and drug response; (iii) AWESOME database to explore SNP effect on phosphorylation, ubiquitination, methylation, and sumoylation sites; and (iv) Ldlink API database of SNP linkage disequilibrium to find out how detected SNPs are linked to other SNPs.
SNPector uses different libraries to import, read, and read data and results. os library is used to run BLAST bash script:
import os
time is used to calculate the time that program.
import time
re refers to regular expression. This library sorts and splits input data with function re.split().
import re
itemgetter module is used to sort BLAST data according to identity, mismatch, and p-value.
from operator import itemgetter
From sys library we used sys.argv[] to convert script to command line, which can be run and controlled from the terminal.
import sys
Then we import the Scripts package to visualize the data as follows:
from Scripts.Circos import DrawCircos from Scripts.Network import DrawNetwork from Scripts.Run_BLAST import RunBLAST from Scripts.Extraction import ExtractSNP from Scripts.APIcommands import APIcommands from Scripts.Visualizations import visualization from Scripts.DataVisualization.DownloadWithAPI.LDmatrix import LDmatrix from Scripts.DataVisualization.DownloadWithAPI.LDhap import LDhap from Scripts.DataVisualization.DownloadWithAPI.LDproxy import LDproxy from Scripts.DataVisualization.CompleteScripts.Ready.ContourPlotWithSeaborn import CounterPlot from Scripts.DataVisualization.CompleteScripts.Ready.CustomLinearRegressionFitSeaborn import LinearReg from Scripts.DataVisualization.CompleteScripts.Ready.CustomLollipopPlot import Lollipop from Scripts.DataVisualization.CompleteScripts.Ready.DendrogramWithHeatmapAndColouredLeaves import DendoWithHeatMap from Scripts.DataVisualization.CompleteScripts.Ready.DensityPlotWithMatplotlib import DenistyPlot from Scripts.DataVisualization.CompleteScripts.Ready.HistogramWithBoxPlot import HistWithBoxPlot from Scripts.DataVisualization.CompleteScripts.Ready.MarginalPlotWithSeaborn import MarginalPlot from Scripts.DataVisualization.CompleteScripts.Ready.ThreeDscatterplot import ThreeDimPlot from Scripts.DataVisualization.CompleteScripts.Ready.UseNormalizationOnSeabornHeatmap import SeabornHeatMap from Scripts.DataVisualization.CompleteScripts.Ready.AnnotatedHeatMap import AnnoHeatMap from Scripts.DataVisualization.CompleteScripts.Ready.NumericalSemantics import NumSChem from Scripts.DataVisualization.CompleteScripts.Ready.ThreeDscatterplot import ThreeDimPlot from Scripts.DataVisualization.CompleteScripts.Ready.VolcanoLD import VolLD
To sort data between the given sequence, Clinvar, AWESOME, BLAST, and PharmGKB, we implement the SNPector variables. This inherits the built-in function open() and nine variables are created as follows:
PharmGKB: data frame describes variant ID, gene name, type of effect, level of evidence, chemicals used to treat the phenotype, and phenotypes;
BLAST_RESULT: data frame lists BLAST output results of alignment of the given sequence against the human genome;
AwesomeDB: data frame lists SNPs chromosome, location, and properties, such as phosphorylation, ubiquitination, methylation, and sumoylation sites;
NCBIclinVar: data frame of SNPs that has clinical impact and involvement in disease;
SNPinDetails: data frame that lists the detected SNPs that SNPector found in the given FASTA sequence;
SNPinDetailsPharmGKB: data frame that lists detected SNPs and its impact on disease development and drug response;
SNPinDetailsAwesome: data frame that lists the properties of detected SNPs;
BLASTfile: function to open and read BLAST output results;
SeqFile: function to read the input file containing the sequence.
Each imported dataset can be found in Extended data21.
RunBLAST() takes the file path of FASTA sequence and starts to align the sequence against the human genome and writes the results to BLAST_RESULT.txt (Extended data: File 221).
os.system('./Scripts/blastn -query GivenSequence.fasta -db ./Data/Hum_Genom38 -outfmt 6 - out ./RESULTS/BLAST_RESULT.txt')
ExtractSNP() reads BLAST_RESULT.txt and sort its with itemgetter() according to the identity, length and p-value, then stores the start and end input given sequence (the query) and subject to use later in the extraction step. It also reads the input FASTA sequence file and stores the sequence variable to use in the comparing extraction step. SNPector provides two inspection modes that can be determined from the terminal, Search and Scan. If mode was “-modesearch”, then SNPector begins to extract all SNPs within the query start and end regardless of their existence in the query. In the mode “-modescan”, SNPector will extract only SNPs that exist in the query
SNPector begins to obtain the alternative nucleotides of SNP through the input sequence and obtains the nucleotides that range from SNP position in ClinVar minus the end position of the subject, to the SNP position in ClinVar minus the end position of the subject plus alternative SNP length to ensure the capture of SNPs from the given sequence and also to detect variants with length more than one nucleotide, finally storing it in the “query_nuc_alt” variable.
query_nuc_alt = sequence[int(snp_pos) - int(subject_end):int(snp_pos) - int(subject_end) + len(snp_alt)]
After the process of extraction the result saved to: “FromAwesom.tsv” file (Extended data: File 321), in which SNPector list all other SNPs that have the same effect in different sites in proteins; “FromNCBI.tsv” (Extended data: File 421), which is list of the SNPs that SNPector detects in a given sequence and retrieves from NCBI ClinVar Dataset; “FromPharmGKB.tsv” (Extended data: File 521), which lista the effect of SNPs in disease development and drug response.
APIcommands() imports SNP IDs from “FromNCBI.tsv” and uses Ldlink API to download “LDhap.csv” file (Extended data: File 621), which describes the allele frequency of extracted SNPs, “LDmatrix.csv” file (Extended data: File 721), which shows how far detected SNPs are linked to other SNPs, and a file titles with the SNP id (e.g. rs516316.csv) (Extended data: File 821), which includes additional information, such as minor allele frequency, linkage disequilibrium and distance of other SNPs linked to the detected SNP.
LDmatrix('./RESULTS/FromNCBI.tsv') LDhap('./RESULTS/FromNCBI.tsv') LDproxy('./RESULTS/FromNCBI.tsv')
DrawCircos() uses SNP properties from “FromAwesom.tsv” file (Extended data: File 321) and searches for other SNPs that have the same properties. SNPector then imports pycircos package to draw SNP location on Circos (Figure 3A).
import pycircos
DrawNetwork() draw network using “FromPharmGKB.tsv” (Extended data: File 521) to get the gene name (e.g. EGFR) and by gene name get all SNP that occur in this gene. Using SNP IDs, SNPector obtains disease names caused by these SNPs, and with the disease name SNPector can extract drugs used in treatment for this disease. Finally, with the drug name SNPector can obtain the clinical annotation of the drug. SNPector uses webweb package to draw the network and export it to .html file (Extended data: File 10).
from webweb import web edge_list = Network Web(edge_list).save("./RESULTS/%sVarPhenoDrugNetwork.html" % GeneName)
Visualization() uses data downloaded in the “LDmatrix.csv” file (Extended data: File 721), and the SNP ID file (e.g. rs516316.csv) (Extended data: File 821) to draw other figures (Figures 3B–K).
SNPector can collect and retrieve information from the user-provided DNA sequence in the simplest way possible. By integrating different databases into SNPector, it is possible to detect the fluctuations in the abundance of SNPs in query through comparison with known variants of human genome. Such steps are accompanied by the use of online and verified sources to gather previously published details regarding target genomic regions and to generate highly informative visualizations of the recovered information.
Many tools, however, provide SNPs annotation, but they are still limited to the information provided (Table 1). SNPector, on the other hand, provides a new technique that extracts SNP from a naked sequence with no prior information. In addition, another benefit of SNPector is to annotate the discovered SNPs from information retrieved from various known databases.
One of the currently growing medical research paradigms is the diagnosis of genetic virulence that accumulates in our genome causing catastrophic health problems. Detection and diagnosis of genetic variation through skill-less computational tools would help researchers reducing the severity of such health complications and improving well-tailored therapies using discovered and previously known information.
SNPector provides and detects all available information about the disease-related SNPs in the given query with minimum user-provided information. It connects between different available information and produces various illustrations depicting SNP related diseases and treatment network, linked disequilibrium, minor allele frequency, similar SNPs with the same mutation effect and other information.
Source code available from GitHub: https://github.com/peterhabib/SNPector
Archived source code as at time of publication: http://doi.org/10.5281/zenodo.355839322.
License: MIT
Homo sapiens chromosome 7, GRCh38.p13 Primary Assembly, Accession number NC_000007.14: https://www.ncbi.nlm.nih.gov/nuccore/NC_000007.14?report=fasta&from=55019017&to=55211628
Zenodo: SNPector Supplementary Data, http://doi.org/10.5281/zenodo.356979021.
This project contains the following extended data:
- Supplementary Files 1–10: output files from SNPector for the FASTA sequence use case (NC_000007.14).
Data are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)