Keywords
Hilsa, anadromous, Bay of Bengal, whole genome, SNP
This article is included in the Genomics and Genetics gateway.
Hilsa, anadromous, Bay of Bengal, whole genome, SNP
Hilsa shad (Tenualosa ilisha) is a migratory fish of the Clupeidae family. It is distributed from the South China Sea and through the Bay of Bengal to the Persian Gulf. The riverine habitats of this fish include the Padma, Jamuna, Meghna, Karnaphuli and the coastal rivers of Bangladesh, the Tigris and Euphrates of Iran and Iraq, the Indus of Pakistan, the rivers of Eastern and Western India and the Irrawaddy of Myanmar (Freyhof, 2014; Pillay & Rosa, 1963). Ecologically, three different types of hilsa shad have been recognized in Bangladesh waters such as anadromous, potamodromous and marine (Milton, 2010).
Hilsa is the most popular and economically important food fish in Bangladesh, contributing 12% of the total fish production and 1.15% of GDP. Of its world catch, 60% amounting to 0.5 million metric tons, comes from Bangladesh (DoF, 2018). Though the overall production of hilsa increased over the years, gross decline in productions were evident from inland waters. A number of factors such as overexploitation, siltation in river beds, decrease in water flow from upstream and fragmentation of the rivers are attributed to this fluctuation in productivity (Ahsan et al., 2014). To enhance hilsa production, programs like the establishment of sanctuaries, restrictions on the use of fishing equipment and a ban on fishing in certain periods of the year to protect parent and juvenile fish have been initiated. It is, however, very important that the management activities be matched with the biological features of the fish for their effectiveness. Inconclusive information about the management units and the level of connectivity amongst them is considered as the major constraint in formulating appropriate hilsa management plans.
There are controversies regarding the number of hilsa stocks in Bangladesh waters. Studies involving morphological and genetic analyses using allozyme, Random Amplification of Polymorphic DNA (RAPD) and mtDNA-restriction fragment length polymorphism (RFLP) markers proved to be insufficient in resolving the stock disputes of this species (Ahmed et al., 2004; Mazumder & Alam, 2009; Salini et al., 2004). DNA markers derived from whole genome sequencing are more efficient to define management units, quantify the extent of adaptive divergence and connectivity among stocks, and to perform mixed-stock analysis (Martinez Barrio et al., 2016; Figueras et al., 2016; Machado et al., 2018). Single nucleotide polymorphic (SNP) markers allow whole genome coverage and high levels of automation. Conventionally, SNP markers are developed by comparing nucleotide sequences with a reference genome. Recent advancement in generating SNPs from reference-free whole genome sequences accelerated identification of SNPs from non-model organisms. Although hilsa is a very important fish biologically and economically, it lacks a reference genome and genomic resources, imposing a severe bottleneck to understand its physiological and ecological requirements. Therefore, we performed whole genome sequencing (Mollah et al., 2018), constructed a draft genome assembly and identified SNPs from T. ilisha of the Bay of Bengal.
We captured ten T. ilisha specimens from the seashore of the Bay of Bengal (21.981753 N 90.305556 E) (Figure 1). All efforts were made to ameliorate harm to the fish by using a seine net of appropriate mesh size (20 mm) to avoid any physical injuries and suffocation. Due to the nature of this species, the fish died immediately after taking them out of the water. Dorsal and caudal fin tissues were immediately clipped on board from a dead female fish (560.42g) and preserved in 96% ethanol. The fish were handled according to the guidelines of the Animal Welfare and Ethical Committee (AWEC) of Bangladesh Agricultural University. The genomic DNA was isolated using the standard phenol:chlorofom:isoamyl alcohol method (Sambrook & Russell, 2001). DNA purity was evaluated using a NanoDrop 2000 Spectrophotometer (ThermoFisher Scientific, cat # ND-2000) and 0.8% agarose gel electrophoresis. DNA was quantified using Qubit 2.0 Fluorometer and Qubit dsDNA HS Assay Kit (ThermoFisher Scientific, Cat. # Q32851) and used to systematically generate the whole genome sequence data (Figure 2).
Photograph of a T. ilisha specimen (a) and a map of Bangladesh showing the sampling site (21.981753 N 90.305556 E) (b).
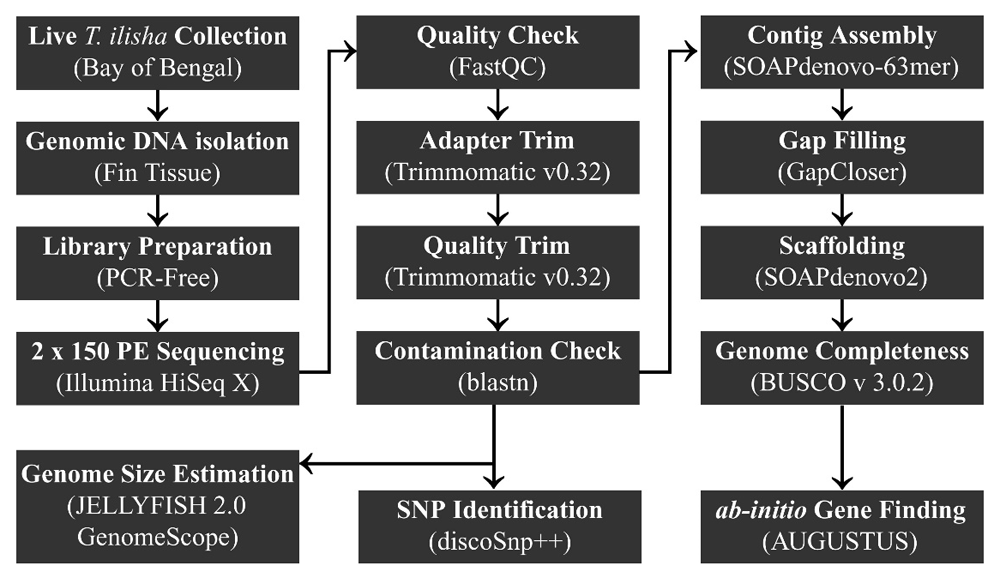
Schematic diagram illustrating the methodology of whole genome sequencing, de novo assembly and identification of SNPs in T. ilisha from the Bay of Bengal.
For sequencing, a PCR-free DNA library was prepared using the Illumina TruSeq DNA PCR-free Library Preparation Kit (Cat. # 20015963), following the manufacturer’s recommendations (Illumina, CA, USA). The library was fragmented, size was selected following the 350 bp insert size scheme and validated using TapeStation (2100 Bioanalyzer,Agilent Technologies, CA, USA). The DNA library was quantified using a Qubit 2.0 Fluorometer as well as by real time PCR (ABI 7500, Applied Biosystems, CA, USA) using the KAPA Library Quantification Kit (Cat. # KK4824) following the manufacturer’s standard protocol with the primer pair Primer 1: 5'-AAT GAT ACG GCG ACC ACC GA-3' Primer 2: 5'-CAA GCA GAA GAC GGC ATA CGA-3'. The PCR condition was followed as initial denaturation at 95°C for 5 min followed by 35 cycles (denaturation at 95°C for 30 sec, annealing/extension/data acquisition at 60°C for 45 sec) and melt curve analysis at 65 – 95°C. Sequencing was performed on the Illumina HiSeqX instrument according to the manufacturer’s instructions. The library was sequenced using a 2× 150 paired-end (PE) configuration (GENEWIZ, LLC. South Plainfield, NJ, USA).
The raw reads were filtered based on quality and length using Trimmomatic-0.32 (Bolger et al., 2014) after evaluating with FastQC v. 0.11.8 (Andrews, 2018) as follows: i) removal of adaptor sequences; ii) removal of read pairs from either ends when the base quality was <20; iii) trimming low quality fragments at both ends of the reads within a window size of 4 bp and an average quality threshold of 15; iv) removal of read pairs having <75 nucleotides. Jellyfish v. 2.2.6 (Marçais & Kingsford, 2011) was used to obtain a separate frequency distribution of three different high occurring kmers (21, 31 and 33) in the raw HiSeq sequence reads, and the histograms were uploaded to GenomeScope for estimating genome size, repeat content, repeat length, unique length and heterozygosity following kmer-based statistical approaches (Vurture et al., 2017).
We assembled the short reads using SOAPdenovo2 genome assembler (Luo et al., 2012), developed specifically for use with next-generation short-read sequences. SOAPdenovo2 uses the de Bruijn graph algorithm. We tested several kmers to assemble the T. ilisha genome and finally selected the assembly with a kmer of 33. The completeness of the T. ilisha genome assembly was evaluated by BUSCO (Benchmarking Universal Single Copy Orthologs) (Simão et al., 2015). For BUSCO analysis (-m geno –sp zebrafish settings), the genome was searched against the Actinopterygii database (actinopterygii_odb9), which was constructed from 20 fish species consisting of 4584 orthologs. AUGUSTUS ab initio gene prediction was performed to predict protein-coding genes. The protein sequences of fish species and other vertebrates, including Rhincodon typus, Cyprinus carpio, Takifugu rubripes, Salmo salar, Mus musculus and Homo sapiens, were downloaded from the NCBI non-redundant protein sequences (nr) database (Table 3) and aligned against the T. ilisha genome using BLASTP (Altschul et al., 1997).
We used discoSnp++ v2.2.10 (Uricaru et al., 2015) with default parameters for reference-free detection of isolated SNPs (SNPs not flanked by other SNPs, Indels or structural variants) by calling SNPs directly from sequence reads without a reference genome. This method identifies isolated SNPs from the sequences of two homologous chromosomes within a single individual.
The estimated haploid genome size of T. ilisha ranged from 649.48 to 660.73 Mb. We observed heterozygosity and a repeat peak (Figure 3), with an estimated heterozygosity of 0.579 to 0.660% and repeats of 8.30 to 13.57% (Table 1). We assembled the draft genome of 710.28 Mb, having an N50 scaffold length of 64157 bp and a GC content of ~43% (Table 2). The whole genome assembly of a notable Clupeid fish, the Atlantic herring, based on short reads (170 bp to 20 kb inserts) was 808 Mb with a scaffold N50 of 1.84 Mb and GC content of 44%, with repetitive elements making up 31% of the assembly (Martinez Barrio et al., 2016). The genome size of another important Clupeid fish, the European sardine (Sardina pilchardus), was estimated to be 655-850 Mb (Machado et al., 2018) and 935-950Mb (Louro et al., 2018).
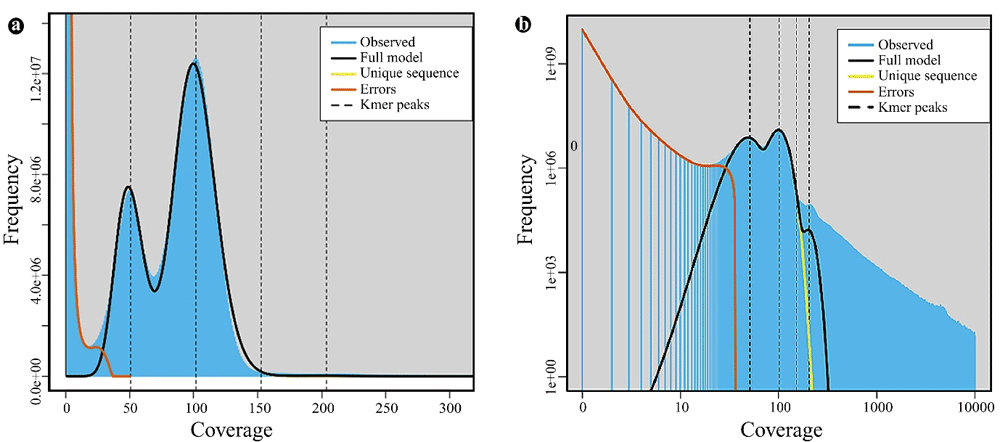
Dataset show the fit of the GenomeScope model (black) based on 33-kmers in Illumina HiSeq sequence reads, max kmer coverage at 300× (a) and 10000× coverage (b).
| Species | No. of matches | % match |
|---|---|---|
| Rhincodon typus | 27062 | 72.26 |
| Cyprinus carpio | 28373 | 75.76 |
| Takifugu rubripes | 28325 | 75.63 |
| Salmo salar | 29339 | 78.34 |
| Mus musculus | 26480 | 70.71 |
| Homo sapiens | 27497 | 73.42 |
The assembled T. ilisha genome was searched for BUSCO analysis against the Actinopterygii database, consisting of 4,584 orthologs constructed from 20 fish species. We found 3,578 complete (C: 78.1%), 3,456 complete and single-copy (S: 75.4%), 122 complete and duplicated (D: 2.7%), 351 fragmented (F: 7.7%) and 655 missing BUSCOs (M: 14.2%). These results suggest higher completeness of the T. ilisha genome assembly of the Bay of Bengal. The BUSCO analysis of its closely related species, the European sardine, showed 84% genome completeness (Louro et al., 2018). The completeness of genome assembly may depend on the sequencing platform used. For example, 92.3% BUSCO completeness was obtained using only the Illumina reads compared to 94.2% completeness from the Illumina + Nanopore reads in the Murray cod (Maccullochella peelii) (Austin et al., 2017).
AUGUSTUS ab initio gene prediction was performed to predict protein-coding genes. We found 37,450 protein coding genes from the assembled T. ilisha genome (Mollah et al., 2019a). To annotate the proteins, predicted amino acid sequences of T. ilisha were aligned against the NCBI non-redundant protein sequences (nr) database of other vertebrates (Table 3) using BLASTP. Among the five vertebrate genomes compared, a minimum of 70.71% genes of T. ilisha was annotated by Mus musculus and a maximum of 78.34% by Salmo salar (Table 3). The numbers of predicted protein coding genes in two other important Clupeids, the Atlantic herring and the European sardine, were 23,336 and 29,408, respectively (Martinez Barrio et al., 2016; Louro et al., 2018).
We identified a total of 792,939 isolated SNPs in T. ilisha genome, of which 510,251 were transitions and 282,688 were transversions (Table 4) (Mollah et al., 2019b). We also detected 155,574 indels ranging in sizes from 1 to 60 nucleotides (Figure 4). A total of 5.3 million raw SNPs in the Atlantic stock and 5.2 million SNPs in the Baltic stock of the Atlantic herring were detected by Feng et al. (2017). In contrast, Louro et al. (2018) identified a total of 2.3 million filtered heterozygous SNPs in the European sardine. Since there is no high quality reference genome available in T. ilisha, we used discoSNP++ because of its nobility and efficiency in detection of SNPs from unassembled genome sequences. Uricaru et al. (2015) genotyped 384 SNPs out of a total of 312,088 discoSNP++ predicted SNPs, of which 368 (95.8%) were accurately validated in the tick Ixodes ricinus.
| SNPs / indels | Type | Number | % |
|---|---|---|---|
| Total SNPs | - | 792939 | 100 |
| Transitions | A>G | 256209 | 32.31 |
| C>T | 254042 | 32.04 | |
| Transversions | A>C | 75912 | 9.57 |
| A>T | 78469 | 9.90 | |
| C>G | 55810 | 7.04 | |
| G>T | 72497 | 9.14 | |
| Transition : Transversion | 1.8 : 1 | ||
| Number of Indels | 155574 | ||
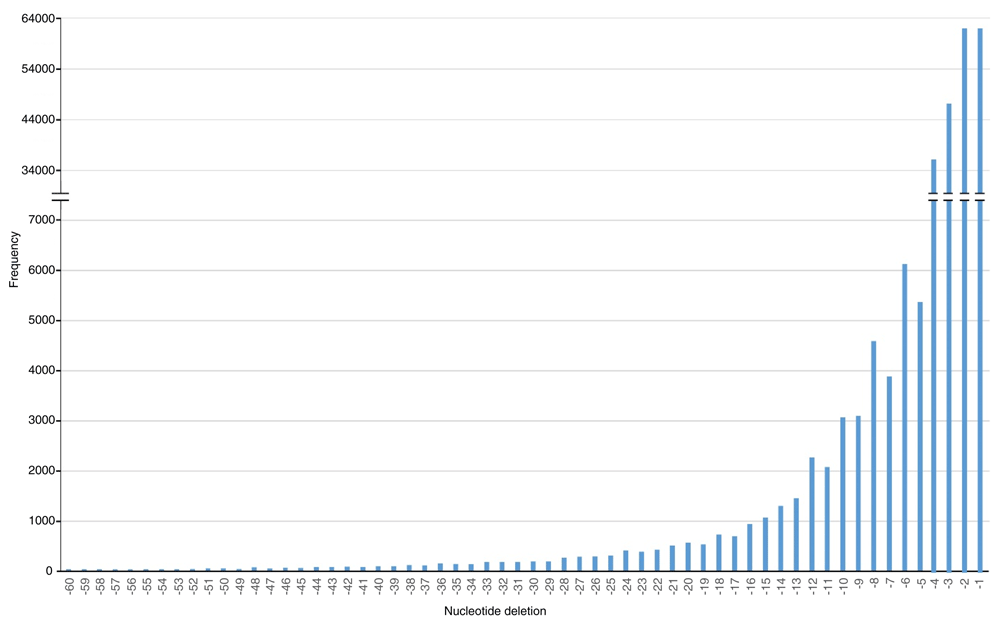
The indel values ranged from 1 to 60 nucleotides. It shows that the frequency of indels decreased with the increase in size.
We report here the first de novo genome of T. ilisha from the Bay of Bengal. The assembled genome can be used as a reference for genetic studies of T. ilisha and related species. The SNPs generated could provide a valuable resource for resolving stock disputes and phylogenetic or adaptation investigation of the Clupeidae family.
Tenualosa ilisha collected from Bay of Bengal, Accession number SAMN07556897: https://www.ncbi.nlm.nih.gov/biosample/?term=SAMN07556897
Tenualosa ilisha whole genome sequencing and assembly, Accession number SRP116260: https://trace.ncbi.nlm.nih.gov/Traces/sra/?study=SRP116260
Tenualosa ilisha whole genome shotgun sequencing project, Accession number SCED00000000: https://www.ncbi.nlm.nih.gov/nuccore/SCED00000000
Zenodo: Amino acid sequences of the proteins predicted from the whole genome of hilsa shad (Tenualosa ilisha) of the Bay of Bengal. https://doi.org/10.5281/ZENODO.2539223 (Mollah et al., 2019a)
Zenodo: Single Nucleotide Polymorphisms (SNPs) identified from the whole genome sequences of hilsa shad (Tenualosa ilisha) of the Bay of Bengal. https://doi.org/10.5281/ZENODO.2538155 (Mollah et al., 2019b)
Data are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
But, the progress of sequences assembly doesn't represent.
I would like to see more information on ... Continue reading This study aim to genome assembly of hilsa shad. these sequences are assembled by SOAPdenovo2.
But, the progress of sequences assembly doesn't represent.
I would like to see more information on the series of the assembly procedure on adequate explanation or explicit mean.
But, the progress of sequences assembly doesn't represent.
I would like to see more information on the series of the assembly procedure on adequate explanation or explicit mean.