Keywords
Gaucher disease, lysosomal storage disorders, cytopenia, β-glucocerebrosidase.
Gaucher disease, lysosomal storage disorders, cytopenia, β-glucocerebrosidase.
Lysosomal deposit disorders constitute a rare group of recessive hereditary monogenic diseases, resulting from the abnormal accumulation of non-degraded material in the lysosomes of different cells of the organism, as a result of the total or partial functional loss of specific lysosomal enzymes or cofactors involved in the degradation of these materials; the resulting lysosomal dysfunction leads to cell dysfunction and clinical anomalies1. The most common lysosomal storage disease is Gaucher disease (GD), with an estimated frequency of 1:40.000 to 1:86.000 inhabitants in the general population, except for the Jewish Ashkenazi ethnic group were it is estimated to be 1:850 births2, characterized by the defective function of the catabolic β-glucocerebrosidase enzyme, which leads to an accumulation of glucocerebroside in the lysosomes of different cells3, causing pancytopenia, hepatosplenomegaly, bone involvement and, sometimes neurological alteration4. The phenotype is variable and there are three clinical subtypes, which are classified by the absence or presence and progression of neurological involvement: type 1 or non-neuropathic form; type 2, the acute neuropathic form of infantile onset; and type 3, the neuronopathic form of juvenile onset5.
In Ecuador, as in Latin America, there are very few reported cases of this clinical entity, and information on this disease is very limited6. In symptomatic patients, GD is usually a progressive disease that, if left untreated, can cause irreversible organ damage, severe morbidity, reduced quality of life and even premature death. Currently there is an effective treatment for GD in enzymatic replacement, which reverses or prevents many of the clinical manifestations of this disorder7.
Despite the availability of accurate and minimally invasive diagnostic tests, patients are often misdiagnosed as a result of the lack of experience with GD and the prolonged delay in accurate diagnosis, and this is likely to be translated into relative rarity of this disease8. Therefore, greater knowledge of the clinical and demographic characteristics of this clinical entity can improve early recognition, reduce the rate of inaccurate or delayed diagnoses for patients with GD, allowing timely treatment aimed at reducing morbidity and preventing irreversible sequels6.
A 29-year-old housewife from Loja-Ecuador, of mixed ethnicity, with no relevant family or personal history, presented to the outpatient service of the Isidro Ayora General Hospital, Loja, Ecuador, in October 2018.
She presented with a pulsating holocraneal headache of slight intensity. Concomitantly, she presented with heartburn which responded positively to self-medication with omeprazole; additionally, she mentioned she was experiencing progressive loss of weight, fatigue and pain in the lower limbs and hands. Physical examination showed ecchymosis and petechiae scattered throughout the body. In the abdomen, hepatosplenomegaly was palpable. Routine laboratory results are summed up in the table below (Table 1).
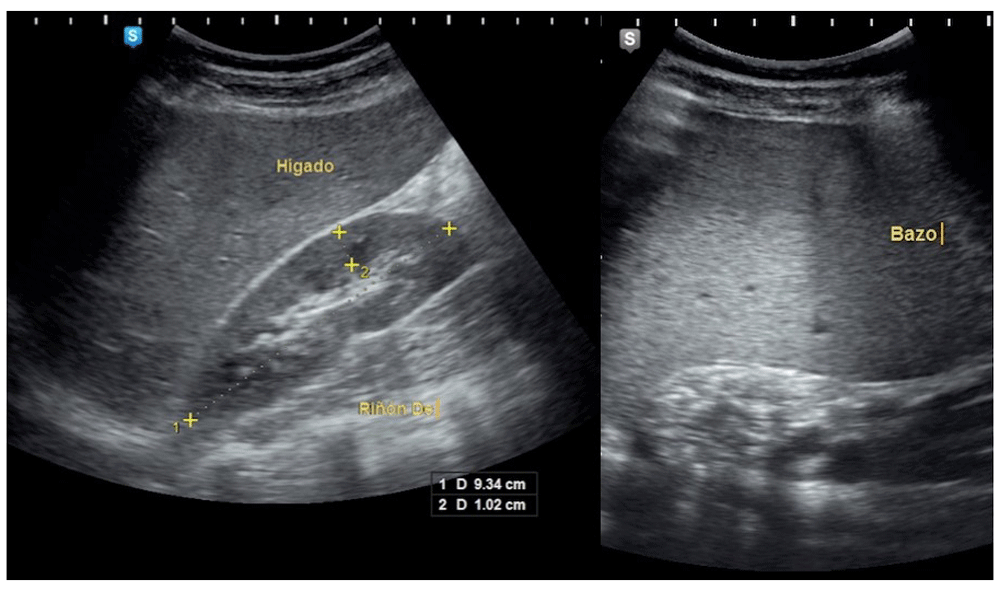
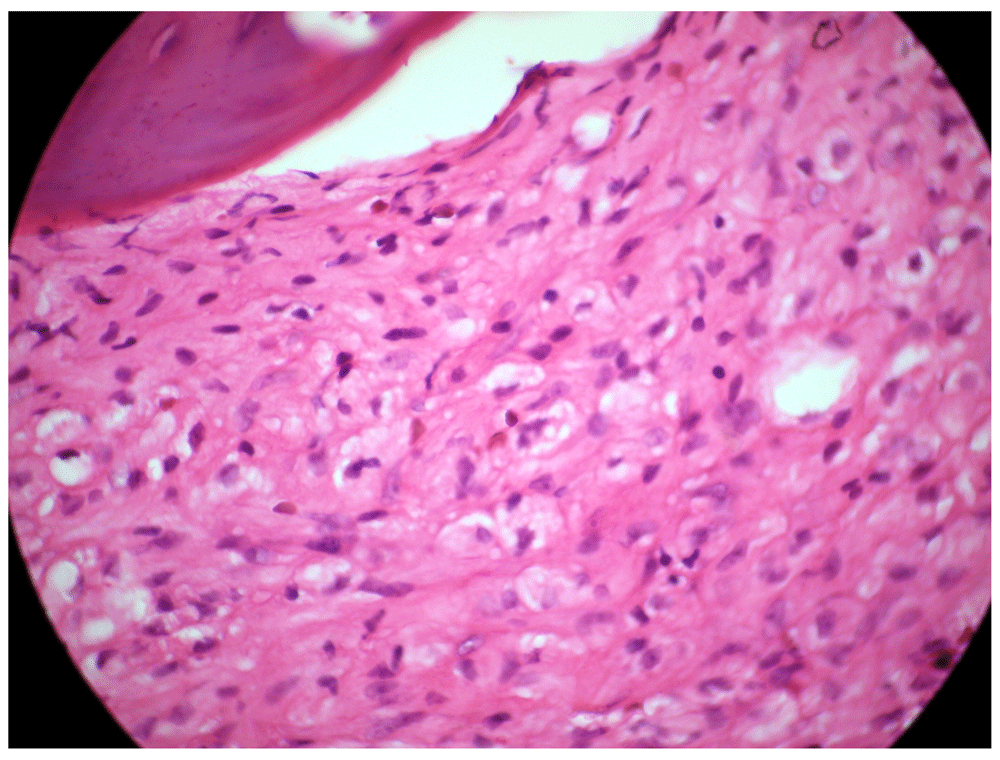
Viral serology tests for hepatitis B, C and human immunodeficiency virus were negative. An ultrasound of the upper abdomen was performed, where there was evidence of a distended liver without occupant injuries and splenomegaly [Figure 1]. Hepatosplenomegaly was observed in the CT-Scan in addition to incipient degenerative osteoarthritis at the dorsal spine. It was decided to perform a bone marrow biopsy, demonstrating abundant cells between the bone trabeculae, consisting of hematopoietic tissue, with a marked decrease in megakaryocytes of hypolobular nuclei, and myeloid-erythroid relationship conserved. In the inter and paratrabecular spaces, abundant clusters of macrophages of the typical broad cytoplasm are observed like “crumpled paper” with small, regular nuclei displaced to the periphery [Figure 2]. The morphological picture is suggestive of lysosomal storage disease, and with the suspicion of GD, quantification of the enzymatic activity of β-glucocerebrosidase was performed, confirming its deficit with a result of 0.27 µmol/L/h (normal range: 2.3 – 12 nmol/h/Ml). Sequential analysis of the GBA gene showed the presence of an apparently homozygous pathogenic alteration in the GBA gene.
Unfortunately, enzymatic replacement could not be performed because Cerazyme (imiglucerase for injection) is not available in Ecuador. Nevertheless, the patient was treated with analgesic (1g of paracetamol generally three times a day) and vitamin supplements (Dayamineral). Currently the patient is waiting for transfer to a foreign institution. Fortunately, the finding of this disease was incidental, and still does not show serious symptoms. At the time of writing the patient is regularly monitored at the Isidro Ayora General Hospital, Loja until the enzymatic medication can be obtained. Currently, the patient persists in good general conditions, without worsening clinical condition. It should be mentioned that she continues with bicytopenia and hepatosplenomegaly, her conditions are expected to be remit once the enzymatic treatment has been administered.
This clinical case is a representative example of the clinical, biochemical and genetic characteristics of GD type 19, characterized by the variability in the signs, symptoms, severity and progression of the disease10. The most common signs and symptoms are: splenomegaly [95%], hepatomegaly [87%], radiological bone disease [81%], thrombocytopenia [50%], anemia [40%], bone pain [27%]11.
GD is caused by mutations in the GBA gene, located on chromosome 1 (1q21), leading to markedly decreased activity of the lysosomal enzyme, β-glucocerebrosidase, which hydrolyzes the glycolipid glucocerebroside in ceramide and glucose12. In the case studied, it was possible to observe a decrease in the enzymatic activity of β-glucocerebrosidase, in addition to alteration of the gene GBA. The consequence of this deficiency is attributed to the accumulation of glucosilcerebroside in macrophages, inducing its transformation in Gaucher cells, which under optical microscopy are usually presented as enlarged cells with eccentric nuclei and condensed chromatin and cytoplasm with heterogeneous appearance as “crumpled paper”13, similar characteristics to the bone marrow biopsy performed in our patient. In our patient, the basic aspects that guided the diagnosis of GD were the incidental finding of a bicitopenia in a routine laboratory examination, in addition to the presence of unexplained hepatosplenomegaly. Without considering the GD in the differential diagnosis of the patient with this type of symptomatology, the definitive diagnosis can be missed or delayed, because the signs and symptoms frequent other more common conditions, including the malignant neoplasms of hematological origin14. Therefore, it is essential to strengthen the knowledge about GD, allowing to reduce the threshold for diagnostic tests and reduce the rate of erroneous diagnoses, in addition to promoting the earliest start of treatment when it is indicated.
GD is usually diagnosed by demonstrating the characteristic “Gaucher cells” in the bone marrow. Microscopically, these cells show a large size, carrying an eccentric nucleus and a cytoplasm that resembles a wrinkled paper13. However, the presence of this type of histological pattern has occasionally been described in several malignant hematological malignancies15, determination of the enzymatic activity of reduced β-glucocerebrosidase or absence is therefore the gold standard for the diagnosis of all GD variants. In the present study a clearly reduced activity of this enzyme was evident, in addition to an apparently homozygous pathogenic alteration of the GBA gene, allowing us to establish a definitive diagnosis of GD.
GD should be considered in the differential diagnosis of patients with unexplained pancytopenia or hepatosplenomegaly. Early recognition will allow the initiation of enzymatic replacement therapy in order to reduce morbidity and improve the clinical aspects of the patient.
Written informed consent for publication of their clinical details and clinical images was obtained from the patient.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)