Keywords
cancer, genetic testing, copy number variation, gene amplification, oncology, targeted therapy
This article is included in the Python collection.
cancer, genetic testing, copy number variation, gene amplification, oncology, targeted therapy
To avoid confusion with a prior software tool of the same name, the name of the app described in this work was changed from InferCNV to InferAMP and the URL updated to http://inferamp.org. The sole changes to the manuscript are: (1) changes to the tool name throughout, and (2) updating with URLs for new tool online and for the GitHub and Zenodo repositories. A revised figure 1 is also provided as the name of the tool is found twice in the figure text.
See the author's detailed response to the review by Andrew C. Nelson
See the author's detailed response to the review by Oscar Krijgsman
Focal somatic gene copy number changes are a widespread event in tumor evolution1. Although these regions of amplification may be large, encompassing many hundreds of genes, typically only one or a small number of genes within the amplified regions are involved in driving tumor growth. Identification of the key driver genes within recurrent amplicons has led to the approval of some therapies that have changed clinical practice (e.g. anti-ERBB2 agents2); however, targeting other amplified genes such as FGFR family members3,4, EGFR5 or KIT6 has frequently proved disappointing. Nevertheless, even some of the more negative trials include occasional strong responses, indicating that sub-populations of patients with amplification of these oncogenes may experience clinical benefit if they can be identified.
With the goal of individualizing treatment for cancer patients, next-generation sequencing from tumor specimens is becoming widely adopted7. In addition to somatic point mutations, several of these assays report copy number changes in assayed genes. Reports for physicians typically present a list of amplified genes without providing a genomic context, leaving physicians and molecular tumor boards to hypothesize which of the listed genes might be driver genes suitable for therapeutic targeting. Given the poor response rates that have often been observed in clinical studies with amplified genes (compared to targeting genes activated by point mutation or fusion), physicians are often appropriately cautious about deciding whether a reported amplified gene may be actionable. Thus, many patients are spared receiving ineffective therapies, but a subgroup of patients who may experience clinical benefit do not get that opportunity.
Here we provide an easy-to-use web tool for analyzing clinical genomics reports of amplified genes. It determines (1) the likely size of amplified genomic regions, (2) which reported genes are co-amplified and (3) which other cancer-relevant genes that were not evaluated in the assay may also be co-amplified in the specimen.
The primary goals are to allow healthcare professionals to determine whether the amplification region surrounding a particular oncogene is relatively small and lacking in other likely candidate cancer drivers (which may indicate increased likelihood that the analyzed gene is a driver) from larger amplicons with additional candidate driver genes (which would suggest a reduced probability that the reported gene is a driver). The approach was developed to analyze the widely used Foundation One test (329 genes) provided by Foundation Medicine but, by simply editing the target gene list, it can be generalized to tests from other vendors which report copy number variation throughout the genome.
InferAMP is written in Python 2.7 with Flask and implemented as a web service running on the Google App Engine (http://inferamp.org). Additional supplied requirements are (1) the coordinates of genes in the human genome, ‘coordinates.txt’ (hg38, UCSC genome browser), (2) The gene list from the assay of interest, ‘foundationone.txt’ and (3) a file listing genes recurrently altered in cancer from COSMIC8 (retrieved 5/3/2018), ‘cosmic.txt’.
An html page with a single query window allows the user to enter a comma-delimited list of genes reported as being amplified. The entry is passed to the script and parsed into individual gene names. An error check is performed to confirm that all entered gene names correspond to gene names in the genome used. The entered genes are considered to be amplified, while the other genes in the assay are considered to be not amplified.
A simplified schema of how the algorithm works is presented in Figure 1, which depicts a chromosomal region containing 30 genes. Seven cancer-relevant genes are present, five of which are evaluated by the genomic assay (Figure 1A). In this test example, three genes were reported to be amplified (Figure 1B). Running the algorithm identifies these amplified genes (8, 13, 20) as well as the nearby assayed genes that are not reported amplified (4, 27). The algorithm considers all genes located between genes reported as amplified to also be amplified (Figure 1C, red shaded region). Because not every gene is assayed, precisely delineating the boundaries of an amplicon is not possible. To address this, the algorithm determines the nearest non-amplified gene at each end of the amplicon and infers that the genes located up to, but not including that gene may be possibly amplified (Figure 1D). The script then returns an html report page listing the entered genes, the amplicons into which they fall (in many cases, several discrete genes will be consolidated into a single amplicon), and also the other cancer-relevant genes within these regions that may be co-amplified with the reported genes. All genes reported include hyperlinks to that gene’s page on COSMIC.
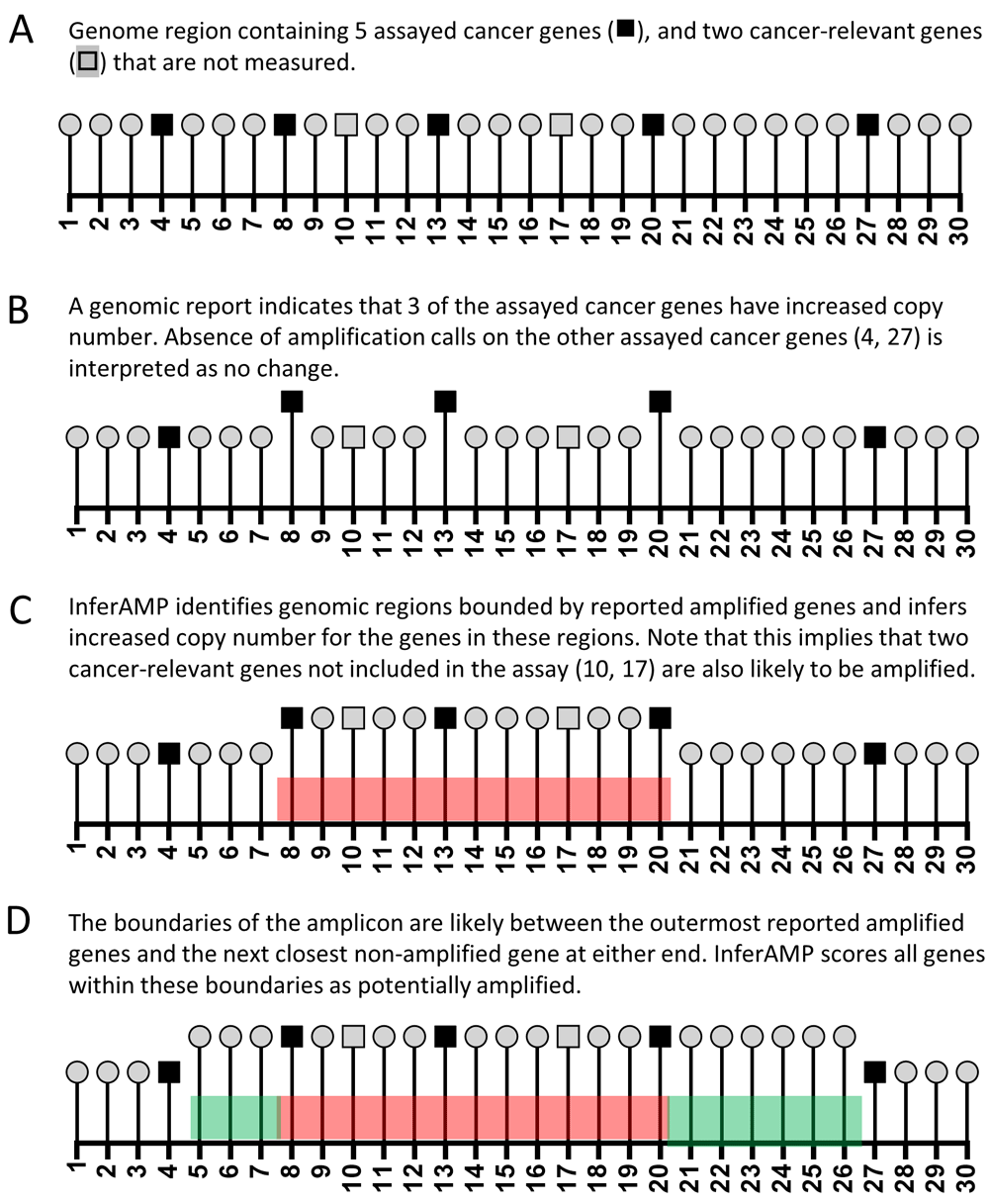
(A). Schematic diagram of a model genomic region with 30 numbered genes, which include a total of 7 cancer-relevant genes. (B) Input scenario for algorithm: a clinical genomics report noting amplification of three genes in this region. (C) Copy number inference for genes in regions bounded by reported amplified genes. (D) Copy number inference for genes surrounding regions bounded by reported amplified genes.
Three use cases taken from genomic reports of patients at our clinic are presented:
A case of esophageal adenocarcinoma with eight reported amplified genes (Table 1), which were resolved by InferAMP to four amplicons. The co-amplification of FGF3, FGF4 and FGF10 with CCND1 (which is likely the driver gene in this amplicon9) might indicate that consideration of FGFR inhibitors may not be helpful if these FGF genes are simply co-amplified passenger genes.
A case of soft tissue sarcoma with five reported amplified genes (Table 2) which were resolved into three amplicons. In the absence of genomic context information, both PDGFRA and KIT might be considered as potentially druggable targets. The demonstration that these are likely co-amplified in a relatively small amplicon might provide further support to this hypothesis. Clinically, both targets are inhibited by imatinib, making joint targeting with a single agent feasible in this case.
The third example is a breast cancer case from our clinic with three reported amplified genes (Table 3). The report highlights one region on chromosome 5, and two regions on chromosome 7. The latter predicted amplicons share a nearby boundary at 7q22.3 suggesting the possibility that there is a regional amplification on 7q encompassing both sets of genes. In this case, MET was judged to be a possible driver amplicon, and the patient had a very strong response to a MET inhibitor10.
We have described a straightforward tool to provide additional genomic context to aid interpretation of amplifications in somatic cancer sequencing reports. Use of this tool may aid decision-making by healthcare professionals about therapeutic options.
The method relies on the accuracy with which test vendors report gene amplification calls. In testing, we identified a small number of cases in which two amplicons were inferred in very close proximity (e.g. Use case 3), which raises the possibility that the assayed gene between the two regions is erroneously not called as amplified. In cases with two or more closely co-located amplicons, users should consider that there is a strong possibility of a regional amplification encompassing both predicted amplicons. Future assays with larger number of genes or more sensitive amplification calling algorithms will likely permit more accurate refining of the boundaries of individual amplicons.
Because the coverage across the genome is somewhat sparse, refining the amplicon boundaries is more challenging than with a more high-density approach like SNP arrays. The primary purpose is to list genes that are potentially co-amplified with a gene identified by a test vendor as possibly actionable in order to allow healthcare professionals to gain further insight into the likelihood that the listed gene is truly the driver gene in that amplicon. Accordingly, we do not distinguish in the report between genes that are likely co-amplified (red genes, Figure 1) from the boundary region genes which are possibly co-amplified (green genes, Figure 1). In any case in which a healthcare professional might consider targeting a non-assayed gene predicted by this algorithm to be amplified (e.g. LIFR11 in Use case 2 and Use case 3), further clinical testing to directly confirm gene amplification would be warranted.
All data underlying the results are available as part of the article and no additional source data are required.
Software available at: http://inferamp.org.
Source code available from: https://github.com/paraickenny/inferAMP.
Archived source code at time of publication: http://doi.org/10.5281/zenodo.324920112.
License: MIT License.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (1)