Keywords
genomics, cancer, immunology, systems biology, R, Shiny
genomics, cancer, immunology, systems biology, R, Shiny
Immuno-oncology (IO) is one of the most promising areas of cancer research, with IO-based treatments demonstrating high efficacy within certain cancer types and subsets of patients1–4. To broaden the utility of these therapies to more patients, fundamental research is required to improve our understanding of tumor-immune interactions—allowing the next-generation of therapeutics and treatment strategies to emerge4. Advances in the IO field are impeded by the inaccessibility of IO study data and results and lack of data standardization, limiting the ability to easily compare results across studies. This has led to the underutilization of existing data, unnecessary study duplication, and failure to achieve rapid consensus in the field5. With the vast increase in the number and scope of IO projects expected in the coming years combined with widespread adoption of genomics and other high dimensional technologies, these problems will be compounded going forward.
We developed the Cancer Research Institute (CRI) iAtlas portal (https://www.cri-iatlas.org) to integrate IO research data, with the goal of providing an interactive, exploratory hub for the IO research community. In doing so, we hope to improve the accessibility and utility of critical resources generated from IO studies. iAtlas is a set of analytic modules—hosted on the web—for studying interactions between tumors and the immune microenvironment. These modules allow researchers to explore associations among a variety of immune characterizations as well as with genomic and clinical phenotypes.
The initial release of iAtlas (April 5, 2018) provided a rich resource to complement analysis results from The Cancer Genome Atlas (TCGA) Research Network on the TCGA data set comprising over 10,000 tumor samples and 33 tumor types6 (“The Immune Landscape of Cancer”; here referred to as “Immune Landscape”). This study identified six immune subtypes that span cancer tissue types and molecular subtypes, and found that these subtypes differ by somatic aberrations, microenvironment, and survival. Per-sample characterizations included total lymphocytic infiltrate (from DNA methylation as well as H&E imaging data), estimated cell type fractions, immune gene signature expression, MHC/HLA type and expression, antigen presentation machinery, T cell and B cell receptor repertoire inference, viral/microbial characterization, associations with pathway disruption and activity, and other analysis results. The Immune Landscape6 manuscript reported on the most novel and potentially therapeutically salient statistical associations between these immune subtypes and the results of the immune characterization. We have continued to develop and evolve the CRI iAtlas application; here, we report the technical design and implementation of iAtlas up to and including the recently released version 1.27. This version includes new features requested by users including: (1) user-defined loading of sample cohorts, (2) a tool for classifying expression data into immune subtypes, and (3) integration of TIL mapping from digital pathology images.
iAtlas is a web-based application to enable data exploration for clinicians, biologists, and informaticists. The inputs and architecture of the application are described below.
The iAtlas app uses structured data and outputs from the Immune Landscape6 study and the TCGA PanCancer Atlas initiative8, which harmonized TCGA data, ensuring uniform quality control and sample inclusion, batch effect detection, normalization across platforms, combination mutation calling from multiple centers, and robustly compiled clinical and outcome data. A key source of iAtlas data is the table summarizing tumor-sample and immune characterizations for 11,080 TCGA patient participants of the TCGA, Table S1 of the Immune Landscape6 manuscript, here termed the “PanImmune Feature Matrix”. Auxiliary data were sourced from files available on this manuscript’s data page at the NCI Genomic Data Commons, from the TCGA PanCancer Atlas Data Mirror, and from the TCGA PanCancer Atlas working space in Synapse (see Data availability). iAtlas data were formatted as data frames (tables) and stored as “feather” files (https://github.com/wesm/feather) on the application server for fast loading (Table 1).
Annotation and browsing of the PanImmune Feature Matrix: iAtlas includes a Data Description page with details on all variables presented in individual modules, with the ability for users to “drill down” on related groups of variables to understand how values were derived. Variables are listed in a text-searchable table containing the name of the variable, the ‘Variable Class’, the unit (if applicable), and whether the variable is numeric or categorical. A ‘Variable Class’ is the name of a group of variables that are of similar type and are often the result of one particular analysis. Clicking on a row exposes a list of all variables in the ‘Variable Class’ and provides links to text descriptions of the analysis methods used to generate the variables.
iAtlas is powered by Shiny9 and makes extensive use of Shiny Modules10 to organize code into composable units (Figure 1). Each iAtlas Analysis module is designed as a Shiny module, allowing simple integration of new analytical functionality. iAtlas uses the tidyverse11 family of R packages (e.g., dplyr12, tidyr13, purrr14, stringr15, tibble16) as well as the wrapr17 package to assist with tidy evaluation. These functions power the data transformations of internal tabular data that are then used to create the interactive plots (i.e., with the plotly18 graphing library) and data tables (via the DT19 wrapper to the DataTables library) seen through the iAtlas modules. We also make heavy use of the crosstalk20 package to enable event-driven updates to the application state. The core iAtlas application is hosted on https://shinyapps.io.
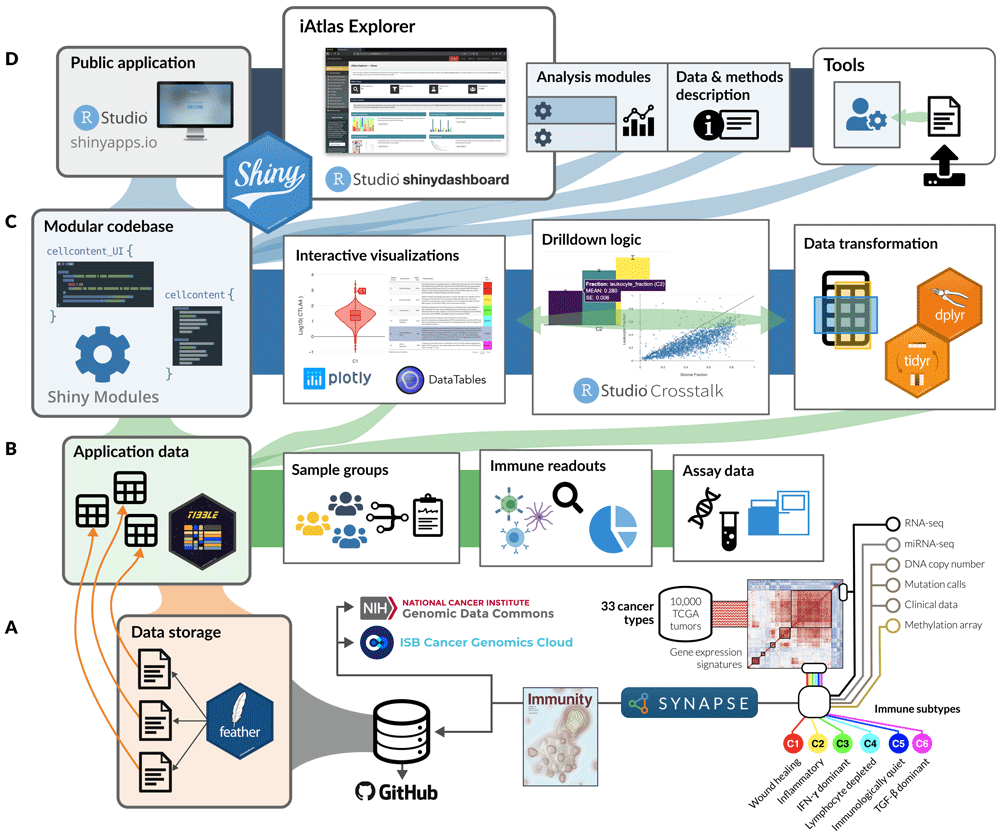
(A) Structured data from immunogenomic analyses, including the Immune Landscape6 study and expanding over time, are organized and stored as flat (i.e., feather) files within Synapse and made available alongside the application code in GitHub. (B) Tabular data from feather files are read from disk into memory to drive all operations related to sample groupings, sample-level immune characterizations (readouts), and more granular—and high dimensional—assay measurements. (C) The core application code is built as a catalog of Shiny Modules, each of which encapsulates logic for data transformation and visualization related to a scientific theme or assay type. (D) Analysis modules, tools, and data description views are hosted in a unified application on shinyapps.io; the layout and connectivity between modules in the iAtlas Explorer space are managed by the shinydashboard21 library.
The main feature of the iAtlas interface is the iAtlas Explorer (Figure 2, found under the EXPLORE tab), which provides several Analysis modules to explore and visualize results. Each module supports a type of exploration, with interactive views and controls to enhance and extend the results and analytics as initially described in the Immune Landscape6 study. The layout of pages and sections within the iAtlas Explorer is driven by the shinydashboard21 package.

A range of Analysis modules (blue boxes above) are available that span from clinical to molecular and imaging data types. Within each module, interactive controls allow researchers to expand views, exposing underlying data and results. Settings are available (green box above) to select the sample groupings (TCGA Study, Disease Subtype, or Immune Subtype) which then propagate through modules.
Within each module in iAtlas, results are displayed relative to Sample Groups, corresponding to defined study cohorts. Several Sample Groups options are pre-loaded in the tool: first, TCGA tumor type (TCGA Study), which are the standard TCGA tumor types collected and designated by the TCGA. Second, TCGA tumor subtypes (TCGA Subtype), a compendium of further subdivision of TCGA studies into molecular subtypes according to publications by the TCGA Research Network22. Finally, a division of tumor samples into distinct patterns of immune response in cancer (Immune Subtypes) is provided6. The choice of Sample Groups is global across all modules but can be updated at any time via the Select Sample Groups element in the side menu. We also allow users to upload custom-grouped samples and analyze those with iAtlas modules. The selection of a sample group defines the samples utilized in all analysis modules. For convenience, group annotations can be displayed in visualizations within each module.
Sample Group Overview: View summary information for user-selected sample cohort groups. There are currently three sections: Custom Groups, Group Key, and Group Overlap. Respectively, these sections permit loading of user-defined sample groups, review of detailed annotations of sample groups in a table, and display of overlap between different types of groupings in a mosaic plot.
Tumor Microenvironment: Explore immune cell proportions in sample groups with two sets of faceted bar charts, one for overall cellular proportions (i.e., leukocyte, stromal, and tumor fraction) and one for computed immune cell proportions (e.g., monocytes, CD8+ T cells, naive B cells).
Immune Feature Trends: Visualize how immune readouts vary across sample groups. Violin or box plots show the distribution of individual values across samples in each group, while heatmaps and scatter plots can be used to explore the correlation between any pair of variables within each group.
Clinical Outcomes: Quantify the relationship between immune response and disease outcome, in terms of either overall survival (OS) or progression free interval (PFI)23. Results are displayed as Kaplan Meier plots as well as heat maps showing the concordance index between variables and survival.
Immunomodulators: Explore the expression of genes coding for immunomodulating proteins6, which include therapeutically important immune checkpoint proteins. Immunomodulators are organized by grouping into three categories: Gene Family (such as “TNF”, “MHC Class II”, “Immunoglobulin”, or “CXC chemokine”), Super Category (such as “Ligand”, “Receptor”, or “Antigen presentation”), and Immune Checkpoint (classified as “Inhibitory” or “Stimulatory”). Violin and box plots are again used to present distributions, and a table provides additional metadata about immunomodulator genes.
Driver Associations: Test and visualize associations between mutations and IO-related response variables. In the Immune Landscape6 study, we reported somatic driver alterations that are correlated with increases or decreases in overall immune cell content, or with the fraction of individual immune cell types. These and other variables can be selected to calculate the significance of relationships in each sample group and view results in a volcano plot.
TIL Maps: We used the results of a recently reported method to assess which spatial regions of hematoxylin and eosin (H&E) whole slide images show evidence of tumor-infiltrating lymphocytes (TILs)24. The method, which uses deep learning, was applied to thousands of H&E slides of the TCGA, allowing slides to be characterized in terms of TIL density and patterns.
Integration with Landscape of IO Drug Target Development: CRI has compiled and published comprehensive overviews describing ongoing immunotherapy drug trials, including targets, agents, and tumor sites and has made summaries available in an online resource, the Immune-Oncology Landscape (IO Landscape ) (www.cancerresearch.org/IO-landscape)25–28. The iAtlas and the IO Landscape resource have been interlinked, enabling researchers to more readily understand the relationship between targeted proteins in IO therapy and the behavior of those targets in tumor tissue.
In IO Target Gene Expression Distributions, the distribution of gene expression values for the selected IO target, by sample group, is displayed in violin plots. Clicking on the expression distribution (violin plot) of a particular sample group, a histogram of the values is displayed.
The IO Target Annotations section provides a searchable table with IO targets and associated annotations. In the rightmost column, a link is provided to a view of the IO Landscape page, the selected target is highlighted in summary barcharts showing the number of agents and cancer types being studied for that target.
In the opposite direction, clicking on targets in the barcharts in the IO Landscape on CRI web pages brings up the target gene expression in iAtlas.
iAtlas Tools are accessible via the TOOLS tab on the iAtlas Portal. Modules in this space of the portal enable users to “bring their own data” for processing through immunogenomic algorithms that drive some of the results presented in the Analysis modules described above.
Immune Subtype Prediction: This tool performs classification of RNA-seq data into one of six immune subtypes as described in the Immune Landscape6 study. Using a new ensemble model based on XGBoost29, researchers can upload their own data for classification30. Each member of the ensemble was trained on a random subset of previously reported immune subtypes6 and features (described below) based on gene expression data from the TCGA PanCancer Atlas Initiative8. All code and methods have been confirmed as reproducible. An R package is available on GitHub (https://github.com/CRI-iAtlas/ImmuneSubtypeClassifier)30.
The submitted expression data—subsetted to the 485 genes that comprised the 5 signatures that produced the immune subtypes—are used to generate robust features of three types: quartiles, binary gene-pairs, and signature-pairs. For example, given a single sample, genes are binned into quartiles and given a bin label (quartile features). Then, similar to the “Top Scoring Pairs” classifier31, genes are paired, and given binary values depending on whether (gi > gj) for two gene expression values, gi and gj. Lastly, signature-pair features are calculated using the five immune subtype signatures, where smn = ∑ij(gim > gjn)/k, where gim is gene i from signature m, gjn is gene j from signature n, and k is the number of gene pairs considered resulting in a value between 0 and 1. The features are computed independently for each sample, and do not require normalization across samples. These features are given to a trained XGBoost classifier which returns a probability of being in any of the six subtypes. Lastly, a “best call” is made with a final trained XGBoost classifier using the six probabilities as input. To validate the robustness of the classifier, TCGA data were processed using four different software pipelines and normalization, showing that classification performance was independent of the gene expression quantification method30. Along with a downloadable table of results, visualizations are also provided. This tool is a convenient way for researchers to apply the methods of the Immune Landscape6 study to their own data without difficult statistical coding.
To use iAtlas, access the web app via https://www.cri-iatlas.org. The software can also be run locally on all platforms (Windows, Mac, Linux). To run the Shiny app locally, a working R installation with necessary libraries is required and an installation of RStudio is recommended.
To install and run the app locally:
One of the initial motivations behind iAtlas was to provide an interactive platform that is able to reproduce figures published in the Immune Landscape6 manuscript but expands that with the ability to generate variations of those figures, for other choices of tumor samples and immune readouts of interest. As an example, in order to reproduce Figure 4A from the Immune Landscape6 publication, which shows the correlation of DNA damage measures with the fraction of leukocytes in the tumor, we began by selecting the EXPLORE tab. We then opened the Immune Feature Trends module and selected the “Immune Subtype” option under Select Sample Groups in the Explorer Settings panel in the left menu. In the ensuing module page, at the Correlations section (Figure 3), we selected the “DNA Alterations” under Select or Search for Variable Class, “Leukocyte Fraction” under Select or Search for Response Variable, and the “Spearman” method under Select or Search for Correlation Method (each a separate dropdown menu). This produced a heatmap identical in content to Figure 4A in the Immune Landscape6 publication. However, the heatmap provides additional information on underlying data via interactivity: by clicking on a heatmap-cell, the underlying data is displayed in a scatterplot. Hovering a cursor over a point in the scatter plot reveals sample-level information.
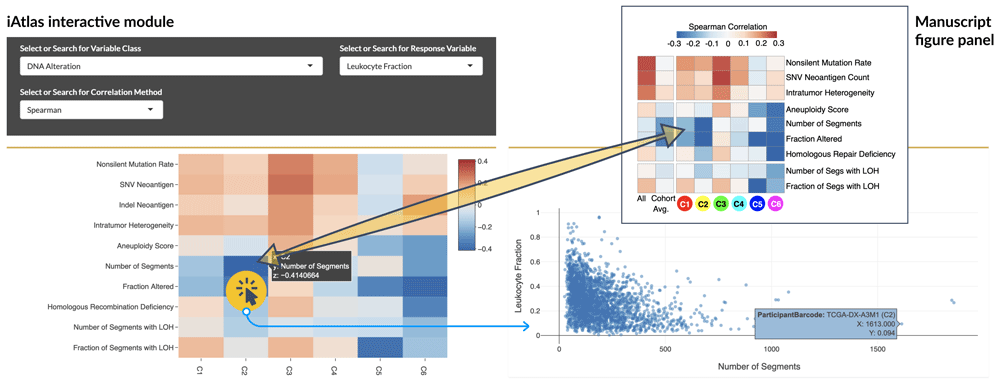
Top right: Original manuscript figure panel from the Immune Landscape6study. Bottom left: Equivalent figure generated in iAtlas. By selecting a specific heatmap cell (highlighted), the underlying data is displayed (Bottom right), using the selections shown. Individual points can be selected to get sample IDs and additional information (blue box).
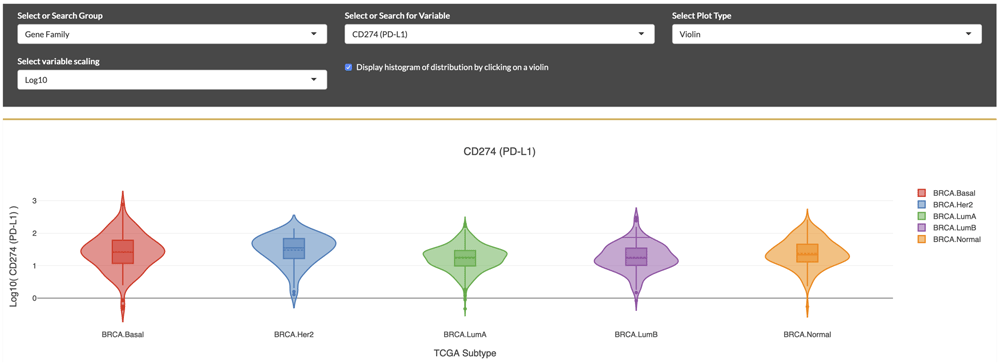
Selection for PD-L1 expression distributions displayed as violin plots within molecular subtypes of breast cancer, according to “PAM-50” classification. Elevated expression is seen in the HER2 subtype.
Table 2 lists the particular manuscript figures (from the Immune Landscape6 publication) that can be reproduced or adapted to specific research questions.
iAtlas Analysis module (Column 1) and examples of figure panels (Column 2) in the Immune Landscape6 manuscript that can be generated using the module. Researchers can use this as a starting point for tailoring figures to their own interests.
With the iAtlas portal, scientists can explore and answer new questions based on specific research interests. For example, we asked: “What is the expression level of PD-L1, a therapeutically important protein, in subtypes of breast cancer?” To answer this question, from the landing page, we first selected the “TCGA Subtype” sample group, followed by the “Breast Invasive Carcinoma (BRCA)” study subset. Next, we selected the Immunomodulators module (Figure 4). Based on a very quick scan of the drop down, we didn’t see any names that matched our gene of interest, so we scrolled further down on the page to view the table of ‘Immunomodulator Annotations’. By typing in the first few letters of a gene name (e.g., “PD...”) into the ‘Search’ field, the table was filtered to a set of matching genes, and we could see that “PD-L1” is the Friendly Name for the gene “CD274” (the approved gene symbol on genenames.org). After returning to the Select or Search for Variable drop down menu above and selecting “CD274 (PD-L1)”, we were able to see a display of violin plots showing the distributions of gene expression across BRCA molecular subtypes. We could then visually compare distributions between subtypes, noticing for example the elevated expression level in the Her2 subtype compared to Basal breast cancer. These comparisons can guide further characterization not only of how gene expression can differ between TCGA subtypes of breast cancer, but also how these subtype-specific differences might correlate with clinical outcomes, as investigated in other studies32–34. Using this module and others, the researcher has the ability to answer new questions which could lead to developments in oncology research.
In order to classify any tumor-derived gene expression samples into immune subtypes6,30, users can select the TOOLS tab (top right), which leads to an interface containing notes, several links and the controls. In order to classify new data, we submitted data as a text file, in this case tab separated, with the first column containing gene IDs and later columns containing samples. A provided example file can be found in the description text. The first row of the data was a header containing sample IDs. Gene IDs can be either HGNC gene symbols (preferred), Entrez ID, or Ensembl identifiers. The locally available data was selected using the Browse button, and the file delimiter was selected, along with gene ID type, using drop down menus. Hitting the GO button produced classifications, signature scores, and cluster probabilities, which were reported in a table that was downloaded as a csv, xlsx, or pdf file. In addition, a barplot with the frequency of predicted subtypes for the submitted data was displayed.
All data required to run the application and describe the Use Cases are available in GitHub and archived with Zenodo7.
CRI iAtlas is a platform that facilitates analysis and exploration of the tumor immune microenvironment by making IO-related data and tools accessible to the research community. iAtlas builds upon the comprehensive TCGA analysis of tumor-immune interactions on 10,000 tumors and illustrates how commonalities and differences of the immune response across 33 tumor types can provide clues for advancing therapeutics. iAtlas provides researchers with the tools to dive deeper into immunogenomic and clinical data and to develop and refine hypotheses regarding tumor-immune interactions that will empower researchers to gain insight and design the next generation of immuno-oncology treatment strategies.
Original data files from the TCGA PanCancer Atlas publication can be found in the NCI Genomic Data Commons (https://gdc.cancer.gov/about-data/publications/panimmune) or the TCGA PanCancer Atlas Data Mirror (https://isb-cancer-genomics-cloud.readthedocs.io/en/latest/sections/PanCancer-Atlas-Mirror.html.
Zenodo: CRI iAtlas (Version 1.2.0). https://doi.org/10.5281/zenodo.39267577.
Folder ‘Data’ contains all data required to run the application and describe Use Cases. This is also available on GitHub.
License: Apache License 2.0.
Source code is available from GitHub: https://github.com/CRI-iAtlas/shiny-iatlas.
Source code for the specific version described at the time of publication: https://github.com/CRI-iAtlas/shiny-iatlas/releases/tag/v1.2.0.
Archived source code at the time of publication: https://doi.org/10.5281/zenodo.39267577.
Hosted iAtlas application on shinyapps.io: https://isb-cgc.shinyapps.io/shiny-iatlas.
Pinned version of the hosted iAtlas app described at the time of publication: https://isb-cgc.shinyapps.io/iatlas_v1-2.
License: Apache License 2.0.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)