Keywords
Embryonic Stem Cells, TDG, Active DNA Demethylation, Base Excision Repair, Neuronal Differentiation, Minigene, Cre/loxP, Tamoxifen
Embryonic Stem Cells, TDG, Active DNA Demethylation, Base Excision Repair, Neuronal Differentiation, Minigene, Cre/loxP, Tamoxifen
In response to the reviewers’ comments and suggestions, the article has been revised as follows:
The integration of the miniTdg transgene in a random genomic location was clarified.
Indicators of statistical significance and extended description of the statistical approaches were added to all figures and respective legends.
Off-target analyses for the two ESC clones generated by CRISPR/ Cas9 were added to the Extended data.
Clarifications and text improvement were made according to the comments and questions raised by the reviewers, referring to the generation and use of the engineered system in cultured cells and animals as well as to technical details.
ypos and ambiguous phrasing were corrected throughout the article.
See the authors' detailed response to the review by Jonathan Sczepanski
See the authors' detailed response to the review by Sebastian Bultmann
See the authors' detailed response to the review by Joseph Torchia
To allow for the differentiation to a multitude of cell types during the development of multicellular organisms, stem cells need to respond to a variety of extrinsic and intrinsic developmental cues and integrate these into epigenetically stabilized gene expression patterns during lineage specification. The underlying mechanisms to ensure this necessary epigenetic plasticity are therefore active and highly dynamic in embryonic stem cells (ESC). One of those mechanisms includes the thymine DNA-glycosylase (TDG) and the downstream factors of DNA base excision repair (BER), which were shown to be involved in the dynamic, locus-specific regulation of DNA methylation. Following the activity of dioxygenases of the ten-eleven-translocation (TET) family that iteratively oxidize 5-methylcytosine (5-mC) to formyl- and carboxylcytosine (5-fC, 5-caC), TDG-induced BER provides a mechanism of active DNA demethylation and, thereby, impact gene regulation (Cortázar et al., 2011; Cortellino et al., 2011; He et al., 2011; Ito et al., 2011; Schuermann et al., 2016; Shen et al., 2013; Weber et al., 2016). Studying the precise function of TDG (and BER) in epigenetic regulation during differentiation of stem cells, however, is challenging due to its multiple and interwoven interactions with other proteins like transcription factors and epigenetic modifiers (Henry et al., 2016; Jacobs & Schär, 2012; Léger et al., 2014), the deregulation of which may cause a plethora of confounding effects. Also, genetic depletion of TDG itself is not compatible with embryonic development (Cortázar et al., 2011; Cortellino et al., 2011). To reduce and circumvent such confounders in functional studies of TDG (and BER) in murine ESCs, we established and validated a Tdg minigene, introduced it into mice and derived a series of different ESC variants. The minigene carries, among other features, a loxP-flanked coding region of Tdg, allowing for a controlled and fast, 4-hydroxytamoxifen (OHT)-inducible depletion of TDG in the background of a homozygous disruption of the endogenous Tdg. By eliminating phenotypic divergence between TDG proficient and deficient ESCs, this versatile model facilitates precise functional and mechanistic investigations into TDG-mediated active DNA demethylation. This novel Tdg minigene introduced into mice as well as ESCs will facilitate future research in the field of active DNA demethylation in vivo and in vitro.
All animal work was carried out in accordance with the Swiss Animal Welfare Act and the guidelines of the Swiss Federal Veterinary Office (SFVO) or with the UK Animals (Scientific Procedures) Act. Housing, breeding and experimentation of mice has been performed with the approval of the Cantonal Veterinary Office of Basel-Stadt (Licenses 10062-H, 1912) or was covered by a project license to Wolf Reik (80/1896) further regulated by the Babraham Institute Animal Welfare, Experimentation, and Ethics Committee. Mice were housed under specific pathogen free condition in individually ventilated cages under 12 h light/dark cycles at 22 ±2°C and 35–65% relative humidity. Animal and colony health was checked daily and quarterly, respectively. Sterilized diet (Kliba-Nafag Extrudate 3436) and water was provided ad libitum.
Tdgtm1Psch mice (http://www.informatics.jax.org/allele/MGI:5487834) were generated from E14 ESC (RRID:CVCL_C320) and backcrossed for more than 10 generations with female C57BL/6JRj mice, obtained at 7-8 weeks of age from Janvier Labs. A Tdg expression cassette (pTCO2-mTDGi.0, Addgene Plasmid #149429) was introduced as single copy into C57BL/6JRj mice by pronuclear injection and crossed into Tdgtm1Psch mice. Genotyping was done by PCR with the HOT FIREPol (Solis BioDyne) on diluted crude DNA preparations (diluted by a factor of five with 10 mM Tris-HCl pH8) by boiling toe clips with 25 mM NaOH/0.2 mM EDTA for 1 h followed by neutralization with 40 mM Tris-HCl. Reactions were performed according to the manufacturer’s recommendation with 35 PCR cycles at 95°C for 30 s, 60°C for 30 s, and at 72°C for 60 s (for details about all used primers and annealing temperatures in PCR reactions, see Extended data Table S1) (Schwarz, 2020).
ESCs were derived from male blastocysts from timed matings of male miniTdgtg/tg//Tdg-/- mice with 2 super-ovulated female Tdg+/- mice and cultured on murine immortalized feeder cells in 2i medium with leukemia inhibitory factor LIF (Merck-Millipore) (Ying et al., 2008).
To introduce the inducible Cre recombinase into the ROSA26 locus, 30 µg of the targeting vector (pROSA26-ERT2CreERT2, Addgene Plasmid #149436) was electroporated into 15×106 mESCs using a gene pulser Xcell (Bio-Rad) at 240 V and 475 μF. Transgenic cells were selected with 8 and 5 µg/ml blasticidin for a week each, before colonies were picked and then amplified without selection pressure. From blasticidin-resistant colonies, genomic DNA was extracted with the “QIamp DNA mini” kit (Qiagen) and screened for a targeted integration at the 3’ junction of the ROSA26 locus by PCR with the Phusion polymerase (NEB) (see Extended data, Table S1) (Schwarz, 2020), applying a general three-step cycling protocol: initial denaturation 95°C for 30 s, 35 times 95°C for 10 s, annealing for 20 s, 72°C for 15–80 s. PCR reactions were analyzed by gel electrophoresis followed by image acquisition with a U:Genius 3 system (Version 3.0.12.0, Syngene).
To evoke a disruption of the NeoR reading frame by CRISPR/Cas9, two guide RNAs ((NeoR)3: 5’-GCCGATCCCATATTGGCTGCAGG-3’, (NeoR)4: 5’-GAAGGCGATGCGCTGCGAATCGG-3’, IDT) were designed and transfected with TransIT-X2 (Mirus Bio) into the TDGiKO1 ESCs. RNP assembly was performed according to the manufacturers protocol (IDT). One day after transfection, single cells were sorted with a FACSaria IIIu (BD BioSciences) into 96-well plates coated with inactivated mouse fibroblasts, based on GFP expression from a co-transfected pEGFP-N1 plasmid (Clontech). Screening for the successful disruption of the NeoR gene was done by PCR as described above and assaying the loss of the neomycin resistance with the Cell Counting Kit-8, according to the manufacturer’s instructions (CCK-8, Dojindo).
ESCs were cultured under a controlled atmosphere (37°C, 5% CO2, 95% humidity) in serum-free 2i medium with 1000 U/ml LIF, without antibiotics unless indicated otherwise. The 4-OHT (Sigma-Aldrich H7904) was dissolved in DMSO (stock 10 mM) and administered at indicated concentrations for 2 h.
Neuronal differentiation was performed as described before (Bibel et al., 2007; Cortázar et al., 2011; Steinacher et al., 2019). Differentiation towards cardiomyocytes was performed by culturing ESCs in classical ESC medium (ESM) with 15% FCS without LIF in non-adhesive petri dishes (Greiner) for at least 10–14 days.
All pictures were taken with a DM IL LED Fluo microscope and MC170 HD camera (Leica) at 100x magnification.
Probes for Southern blotting were generated by PCR amplification with Taq DNA Polymerase (Qiagen) from the ROSA26 targeting plasmid using the DIG DNA labeling mix (Roche). Southern blotting was done with 20 µg of genomic DNA, extracted by the “Genomic tip 100G” kit (Qiagen). Probes were hybridized according to the “DIG Application Manual for Filter Hybridization” by Roche. Signals were acquired by exposing the membrane to chemiluminescence detection films (Amersham/GE Healthcare) and digitalized by a CanoScan 8400F scanner with CanoScan Toolbox (Version 4.9.3).
Excision of the miniTdg gene by Cre was assessed by qPCR with the “Rotor-Gene SYBR Green PCR Kit” (Qiagen) on a Rotor Gene 3000 (Qiagen) system. After 95°C for 5 min, qPCR reactions followed a two-step cycling (40x) protocol: 95°C for 5 s, 60°C for 30 s, terminated by ramping from 60 to 95°C in 175 s to generate a melting curve. PCRs to exclude background recombination by Cre were done with NEB Phusion polymerase as described above.
Total RNA was extracted with the RNAeasy Mini Kit (Qiagen), including on-column DNase digest, and reverse transcribed with RevertAid First Strand cDNA synthesis kit (ThermoFisher) using oligo-dT primers. qPCR was performed as described above with marker-specific primers (see Extended data, Table S1) (Schwarz, 2020).
Protein levels were analyzed by western blotting of 50 µg of NP-40 (or SDS) whole-cell extracts, separated by SDS-PAGE, onto a nitrocellulose membrane (Amersham) and immunodetection with anti-mTDG antibody (rabbit polyclonal L58, Lab P.Schär, dilution 1:10’000, 1 h at 33°C) and anti-GAPDH antibody (Sigma-Aldrich Cat# G9545, RRID:AB_796208, dilution 1:20’000, 1 h at RT). Chemiluminescence signals were detected with a Fusion FX7 system (Software version 16.15.0.0, Vilber).
DNA was extracted and purified with the Genomic tip 100G Kit (Qiagen). High-performance liquid chromatography–tandem mass spectrometry analysis was performed on 10 µg of genomic DNA as described before (Weber et al., 2016).
To facilitate genetic manipulation of the Tdg gene (~28 kb including the putative promoter), we first constructed a synthetic Tdg minigene (miniTdg, ~11 kb) (Figure 1A). The minigene consists of the endogenous Tdg promoter and terminator sequences flanking the Tdg coding sequence (CDS). It also includes the rabbit β-globin intron at the authentic position of the first intron of the Tdg transcript variant 2 (Genebank NM_172552.4). This intron allows for the expression of the two naturally occurring Tdg splice variants (Um et al., 1998). In addition, the miniTdg contains two loxP sites in the same orientation for Cre-recombinase-mediated excision of the coding region. A plasmid carrying the miniTdg gene (pTCO2-mTDGi.0) was introduced into C57BL/6 mice at a random genomic location by pronuclear injection and offspring carrying the minigene were crossed into heterozygous Tdgtm1Psch mutant mice (Kunz et al., 2009). When breeding heterozygous mice for the miniTdg transgene and the Tdg KO allele, the genotypes of offspring followed the expected Mendelian pattern for two loci (Figure 1B). The miniTdg allele segregated with a 3:1 ratio, consistent with a random integration into a single genomic locus. About 25% of born mice with the miniTdg gene were homozygous for the Tdgtm1Psch KO allele while no homozygous Tdg KO mouse was obtained without the transgene. This shows that the miniTdg transgene is fully functional in complementing the developmental defects and lethality of homozygous Tdgtm1PschKO embryos (Cortázar et al., 2011; Cortellino et al., 2011). MiniTdg complemented mice were healthy and fertile with normal lifespan.
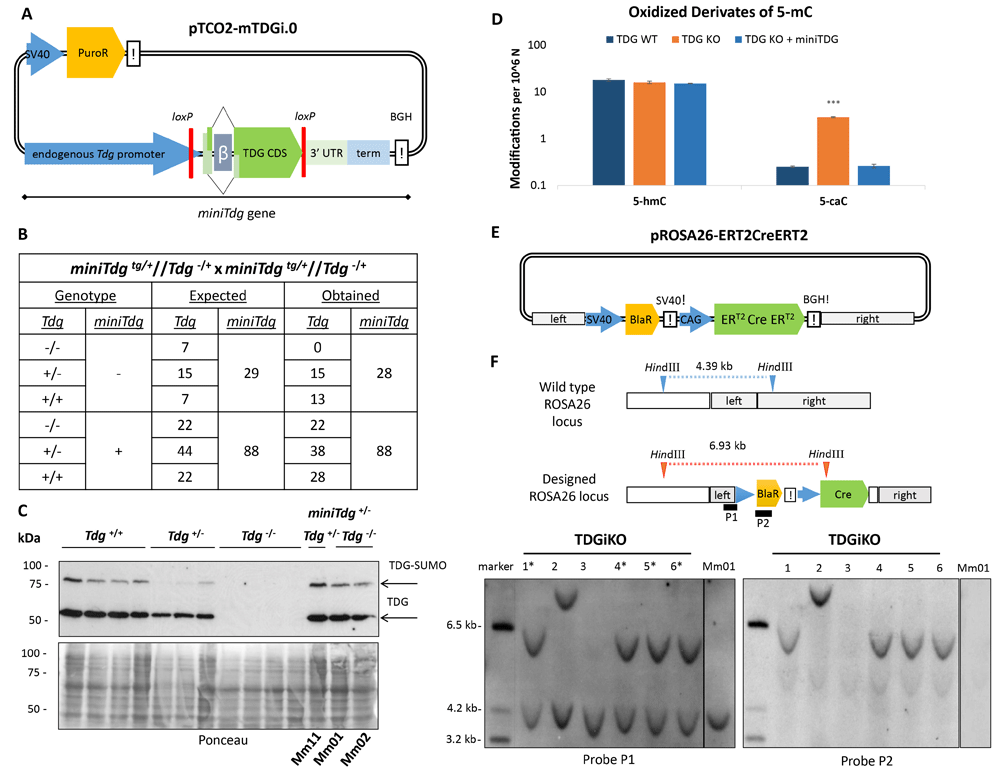
(A) Synthetic miniTdg gene on the plasmid integrated to a random genomic locus by pronuclear injection. miniTdg consisting of 6 kb endogenous Tdg promoter, the Tdg coding sequence (CDS) including a chimeric splice donor (Tdg exon 1 of transcript variant 2 NM_172552.4, rabbit β-globin intron), to allow for alternative splicing, 3 kb of the 3’ untranslated region (3’ UTR) and terminator (term) of Tdg, and the bovine growth hormone terminator sequence (BGH!). loxP sites are indicated in red. (B) Expected and obtained genotype distribution in offspring (n=116) from crosses of heterozygous mice. (C) Top: Immunoblot for TDG of whole-cell SDS-extracts from derived ESCs with different genotype. Bottom: Loading control by Ponceau staining of the membrane. (D) Accumulation of oxidized 5-mC derivates measured by HPLC-MS/MS in ESCs with the indicated genotype. For each genotype, means and standard deviations of two independent clones with 1 or 2 repeated measurements are shown. Asterisks indicate p-values of Student’s t-test, compared to TDG WT: ***p-value < 0.001. (E) Scheme of the targeting construct pROSA26-ERT2CreERT2 for the ROSA26 locus with the tamoxifen-inducible Cre recombinase (Matsuda & Cepko, 2007). ERT2-Cre-ERT2 expression is under the control of the synthetic cytomegalo-virus/chicken-actin/beta-globin promoter (CAG). Homology arms for ROSA26 targeting are indicated in light grey. A blasticidin resistance cassette (BlaR) for positive selection is under the control of the SV40 promoter and terminator. (F) Predicted restriction pattern of ROSA26 WT and integration events (top) and fragments detected by Southern blotting (bottom). Left: The hybridization probe (P1) locating to the left ROSA26 homology arm detected a genomic fragment of 6.9 kb. The 4.4 kb fragment represents the wild-type ROSA26 allele. Right: Detection of possible off-target events using a second probe (P2) locating to the blasticidin selection marker within the targeting construct. See Underlying data for the raw data behind this figure (Schwarz, 2020).
Three ESC populations were then derived from male blastocysts from crosses of miniTdgtg/tg//Tdg-/- with Tdg+/- mice (Extended data, Table S2: Mm11, Mm01, Mm02) (Schwarz, 2020). In these ESCs, TDG protein levels were similar to those in wild-type ESCs (Figure 1C), indicating that the miniTdg gene is regulated as the endogenous Tdg. Furthermore, no silencing of the transgene was noticed over several generations of animal breeding or culturing of ESCs. To assess functional integrity at the molecular level, we measured the accumulation of oxidized 5-methylcytosine (5-mC) derivates, 5-hydroxymethylcytosine (5-hmC) and 5-carboxylcytosine (5-caC), the substrate for excision by TDG (Maiti & Drohat, 2011; Shen et al., 2013) (Figure 1D). Mass spectrometry showed that TDG deficient ESCs accumulated 5-caC as expected, while ESCs expressing TDG solely from the miniTdg gene showed 5-caC levels similar to those in wild-type ESCs.
To allow for a controlled excision of the miniTdg coding region, we introduced a tamoxifen-inducible Cre recombinase expression cassette (Figure 1E) into the non-essential ROSA26 locus (Soriano, 1999). Screening 60 blasticidin-resistant colonies by PCR for targeted integration at 3’ ROSA26 locus yielded six clones (TDGiKO1-6). Southern blotting confirmed correct heterozygous integration of the complete ERT2-Cre-ERT2 cassette (Matsuda & Cepko, 2007) for four of the six clones (Figure 1F, left, asterisks). Potential additional off-target integrations were tested and excluded by hybridizing a second probe within the transgene (Figure 1F, right). Original Southern blotting images are available as Underlying data (Schwarz, 2020).
Taken together, we established a miniTdg gene that complements expression and function of the endogenous Tdg gene in vivo and in vitro. The functional integrity of the transgene is confirmed by rescuing phenotypes of Tdg KO defects at the systemic and cellular level, namely the rescue of embryonic lethality and suppression of 5-caC accumulation. The introduction of a Cre-recombinase to the ROSA26 locus, without off-targeted events, will allow conditional inactivation of the miniTdg in ESCs.
In addition, we generated ESC clones that ease further genetic manipulations involving selectable marker genes. We eliminated the neomycin resistance gene, which was introduced at the endogenous Tdg locus to generate the original Tdg KO allele (Kunz et al., 2009), by applying a CRISPR-guided Cas9 nuclease. Using two guide RNAs targeting the 5’ and 3’ end of the NeoR gene, we generated two ESC clones (TDGiKO1.1 and TDGiKO1.2) that bear a deletion of 776 and 767 bp, respectively, and are no longer resistant to neomycin (Extended data, Figure S1A-C) (Schwarz, 2020).
To make the inducible TDG depletion (Figure 2A) applicable to differentiation experiments that often depend on complex and strict cell culture procedures, we aimed at establishing a short Cre-induction procedure minimizing any interference with standard differentiation conditions. Titrating 4-Hydroxytamoxifen (OHT) and measuring miniTdg excision by quantitative PCR, we identified OHT concentrations of 1-5 µM in serum-free 2i (Figure 2B) and 5-10 µM OHT in serum-containing medium (Figure 2C) to yield maximal excision efficiencies without impairing cell viability (Figure 2D). We note that lower OHT doses (down to 100 nM) for longer duration (up to 48 h) may be applied as well, depending on the experimental procedure and objective. While the miniTdg excision product is readily detectable by PCR in different TDGiKO ESCs after OHT treatment, no unintentional background activity of the Cre recombinase is detectable before OHT induction (Figure 2E). Following the kinetics of TDG depletion by SDS-PAGE/immuno-detection, we found that TDG was reduced below detection limit (<2% of endogenous TDG) 24 h after Cre induction for 2 h (Figure 2F and Extended data, Figure S1D) (Schwarz, 2020).
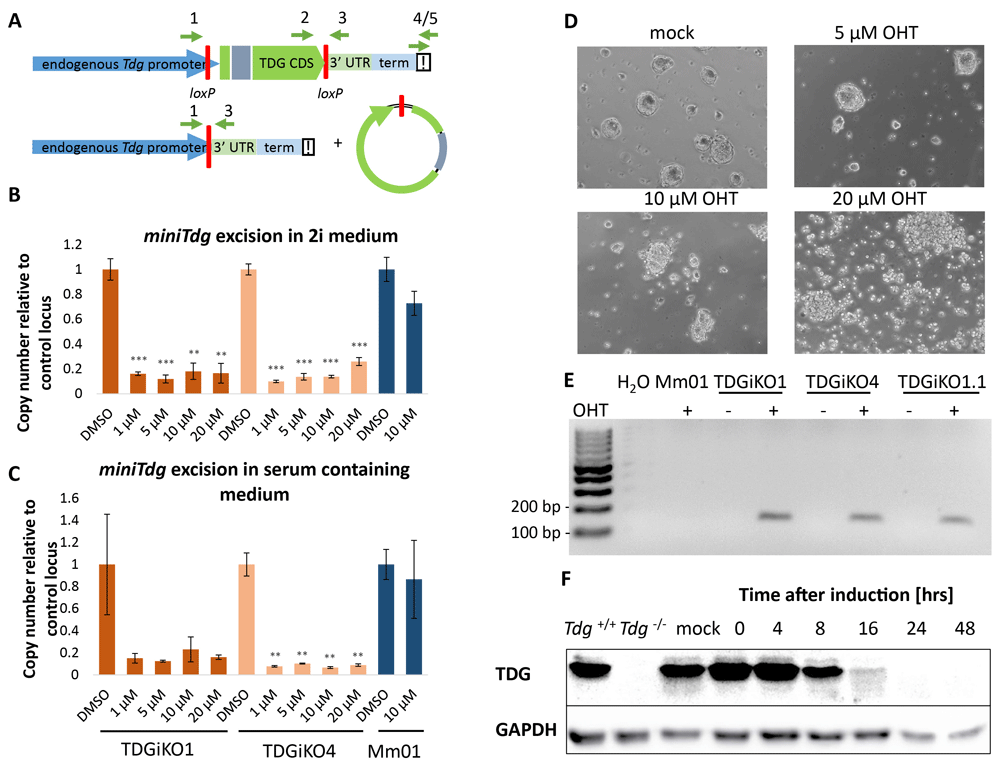
(A) Scheme of the miniTdg cassette before (top) and after (bottom) Cre-mediated recombination. Position of PCR primers are indicated with green arrows. (B) Quantitation of miniTdg excision upon OHT administration in TDGiKO1, TDGiKO4 and parental ESCs without the Cre recombinase (Mm01). Genomic DNA of cells was extracted two days after OHT treatment with indicated concentrations for 2 hours. Copy number of the miniTdg is measured by qPCR using primers 2 and 3, normalized to the nearby terminator region (primers 4/5) and a control locus on chromosome two. Shown are means and standard error from technical quadruplicates per clone. Asterisks indicate p-values of Student’s t-test, compared to DMSO control: ** p-value < 0.01, ***p-value < 0.001. (C) Quantitation of miniTdg excision as in (B), but Cre induction executed in ES medium containing 15% FCS. (D) Phase-contrast images of TDGiKO1 ESCs in 2i medium, two days after OHT treatment for two hours at indicated concentrations. (E) Detection of Cre recombination events (158 bp) by PCR using primers 1 and 3 (A) before and after addition of OHT. (F) Time-course assessment of TDG protein levels expressed from the miniTdg gene. Immunodetection with an anti-TDG antibody was performed with western blotted NP-40 cell extracts of 2i cultivated TDGiKO1 ES cells after Cre induction by 1 µM OHT for 2 h. See Underlying data for the raw data behind this figure (Schwarz, 2020).
We also assessed the tissue specific induction of Cre-mediated miniTdg excision in vivo. We crossed miniTdg mice with mice expressing Cre-recombinase under the control of a FoxN1-promoter (http://www.informatics.jax.org/allele/key/62500) which drives expression in thymic epithelial cells (TECs), but not in thymic cells (TCs). We observed that the mRNA and protein expressed from the miniTdg are reliably depleted in TECs expressing FoxN1-driven Cre, whereas TDG levels are much less affected in TCs (Extended data, Figure S1E/F) (Schwarz, 2020).
These data demonstrate the functionality and tight regulation of the inducible miniTdg KO model in murine ESCs as well as in mice. A short-time Cre induction with 1–5 μM or 5–10 μM OHT (in 2i+LIF or 15% FCS-containing medium, respectively) is sufficient to mediate excision of the miniTdg, resulting in depletion of TDG below detection within 24 hours. This rapid depletion of TDG is presumably facilitated by the cell cycle-regulated proteasomal degradation of TDG via the ubiquitination at its PCNA-interacting peptide (PIP) motif (Hardeland et al., 2007; Shibata et al., 2014).
To validate the molecular phenotype of TDG depletion in our newly derived TDGiKO ESCs (TDGiKO1 ESCs and derived NeoR-deleted TDGiKO1.1), we again assessed changes in levels of oxidized 5-mC derivates, 5-formylcytosine (5-fC) and 5-caC following Cre-mediated miniTdg deletion. One week after induction, we measured a 4.4 and 4.2-fold accumulation of the TDG substrates 5-fC and 5-caC in TDG-depleted TDGiKO ESCs, respectively, but no changes of 5-hydroxymethyl cytosine (5-hmC), which is not recognized by TDG (Figure 3A). This accumulation reflects well the previously reported measurements in constitutive Tdg KO cells (Shen et al., 2013; Steinacher et al., 2019).
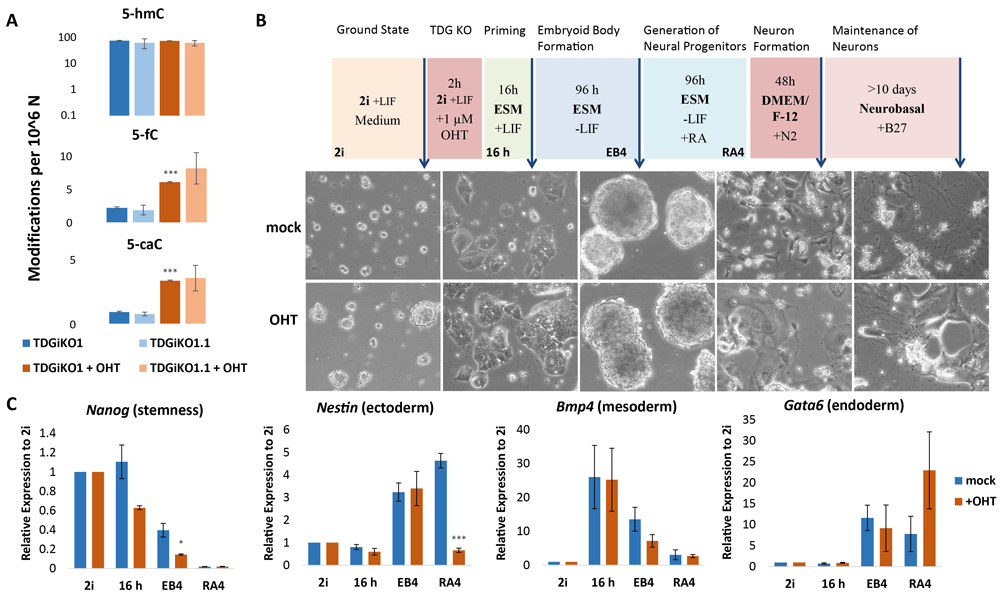
(A) Measurement of oxidized derivates of 5-mC in selected ESCs by HPLC-MS/MS, cultivated in 2i medium + LIF, one week after TDG depletion by 1 µM of OHT for 2 h. Shown are the means and standard deviation of 2-3 biological replica per condition. (B) Scheme of the neural differentiation protocol (Bibel et al., 2007), adapted with a 16 h priming step in ESC medium for ESCs grown in 2i medium. Representative phase-contrast images of TDGiKO1 cells with (+OHT) and without (mock) TDG depletion at the differentiation stages indicated (arrows). (C) Expression of pluripotency and germ layer marker genes were assessed by qRT-PCR in TDGiKO1 cells at the stages of differentiation as marked in (B). Expression was normalized to the housekeeping genes Rps13 and Eef1a1. Shown are means and standard errors of 3 biological replicates. Asterisks indicate p-values of Student’s t-test, compared to the non-induced condition: *p-value < 0.05, ***p-value < 0.001. See Underlying data for the raw data behind this figure (Schwarz, 2020).
Finally, we re-tested the pluripotency and differentiation capacity of some of the engineered TDGiKO ESC clones (Extended data, Table S2) (Schwarz, 2020), applying a protocol for all-trans retinoic acid (RA) induced in vitro differentiation towards the neuronal lineage (Bibel et al., 2007) with minor adaptations. TDG depletion was induced just before an additional culturing period of 16 h in serum containing medium, which promotes the transition from the ground (cultured in 2i medium) to the primed ESC state (Figure 3B). Formation of embryoid bodies and neural progenitor cells was triggered by LIF withdrawal and later the addition of RA, before terminal differentiation was induced by replating the cells in media for neural cells. When subjecting TDG proficient TDGiKO1 ESCs to neural differentiation, we observed an upregulation of the neural marker Nestin (Figure 3B, C) and, ultimately, the formation of neuron-like cells (Figure 3B). As expected during embryoid body (EB) formation, LIF withdrawal resulted in an induction of lineage-specific genes of all three germ layers and the repression of the pluripotency marker gene Nanog. This indicates the ability of the TDG proficient TDGiKO ESCs to commit to cell types of all germ layers. In support of this, continued cultivation of non-induced TDGiKO1 and TDGiKO1.1 in ESM without LIF led to a spontaneous differentiation to cardiomyocytes (mesodermal cell type) forming beating organoids (see Extended data Videos 1 and 2) (Schwarz, 2020). While the early initiation of lineage-specific genes seemed to be unaffected by TDG depletion prior to differentiation, the formation of neuron-like cells (Figure 3B, bottom) was impaired. This deficiency to stably commit to and/or maintain the neuronal lineage was indicated by a low number of neuron-like cells formed and the loss of cells with Nestin expression following RA application. Upon loss of TDG, we furthermore observed a significant difference in the downregulation of the pluripotency marker Nanog as well as and dysregulation of the endodermal marker Gata6 (Figure 3C).
Thus, when subjecting the TDG-depleted TDGiKO ESCs to in vitro differentiation, we obtained data consistent with previous observations in constitutive Tdg KO ESCs (Steinacher et al., 2019). This highlights the usability of the newly established ESCs in TDG and BER-related research, with the advantage of eliminating clonal divergences between TDG proficient and deficient cells. While here, we focused on the reproduction of published Tdg KO phenotypes, the fast and reliable depletion of TDG in the TDGiKO ESC model now provides a tool for in-depth execution point analysis of TDG-BER mediated active DNA demethylation during cell differentiation or in any other context of interest. Furthermore, the loxP sites flanking the entire coding region of the miniTdg gene enable recombinase-mediated cassette exchange to introduce TDG separation of function variants devoid of catalytic function or harbour mutations that ablate posttranslational modifications (Hardeland et al., 2000). Such an exchange with functional TDG variants or fusion proteins is not easily applicable with other available murine conditional Tdg knockout systems (e.g.: Cortellino et al., 2011; Hassan et al., 2017; Hu et al., 2014; Song et al., 2013). Additionally, the availability of mice carrying the miniTdg, which can be combined with Cre-driver mice of interest by crossing, allows the translation of in vitro observations into in vivo settings. All in all, we provide a versatile, well-controlled and validated toolbox (Extended data, Table S2) (Schwarz, 2020) to facilitate functional and mechanistic research on TDG-mediated active DNA demethylation and BER in cell culture and animal models.
Zenodo: Inducible TDG knockout models to study epigenetic regulation. http://doi.org/10.5281/zenodo.4075154 (Schwarz, 2020).
This project contains the following underlying data:
Raw_Data_Figure1.zip. Database excerpt for genotyping of mouse crossings, original pictures of Southern and western blots and MS-quantification of oxidized mC derivates (also for figure 3).
Raw_Data_Figure2.zip. qPCR data of miniTdg excision, original pictures of OHT-treated ESCs and western blots.
Raw_Data_Figure3.zip. Original pictures of differentiating cells and qPCR data of pluripotency and germ line markers.
Raw_Data_Extended_Figure.zip. Sequencing data for NeoR excision and predicted off-targets, and predicted off-targets, absorption values of G418-treated ESCs, qPCR data for miniTdg excision and original pictures of western blots.
Zenodo: Inducible TDG knockout models to study epigenetic regulation. http://doi.org/10.5281/zenodo.4075154 (Schwarz, 2020).
This project contains the following extended data:
Extended Figure and Tables.pdf. Supplementary information as referenced in the text.
Extended_Video_1_TDGiKO1.mp4. Video of “beating bodies” from an ESC clone.
Extended_Video_2_TDGiKO1.2.mp4. Video of “beating bodies” from a second ESC clone.
Data are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)