Keywords
Asgard archaea, 2D, tree of life, LUCA, phylogenomics, nonstationary, rooting, eukaryogenesis
Asgard archaea, 2D, tree of life, LUCA, phylogenomics, nonstationary, rooting, eukaryogenesis
Models of character evolution are essential to determine the evolutionary relationships of organisms. Phylogenetic models that employ protein structural domains as characters place Asgards as sister to other archaea (Figure 1a), and archaea sister to bacteria in the “tree of life” (ToL)1–3. Whereas several analyses that employ amino-acids as characters fail to resolve the archaeal radiation (Figure 1b) or to identify a distinct ancestor of archaea4. Conflicts between different studies that employ different character types are often due to incompatible assumptions about the character-evolution processes5–7. In a recent study, Williams et al.4 compared the performance of several character-evolution models to evaluate which one of the ToL hypotheses is better supported. The authors tested the performance of several substitution models for amino acid characters using empirical data, but models for protein-domain characters with simulated data4.
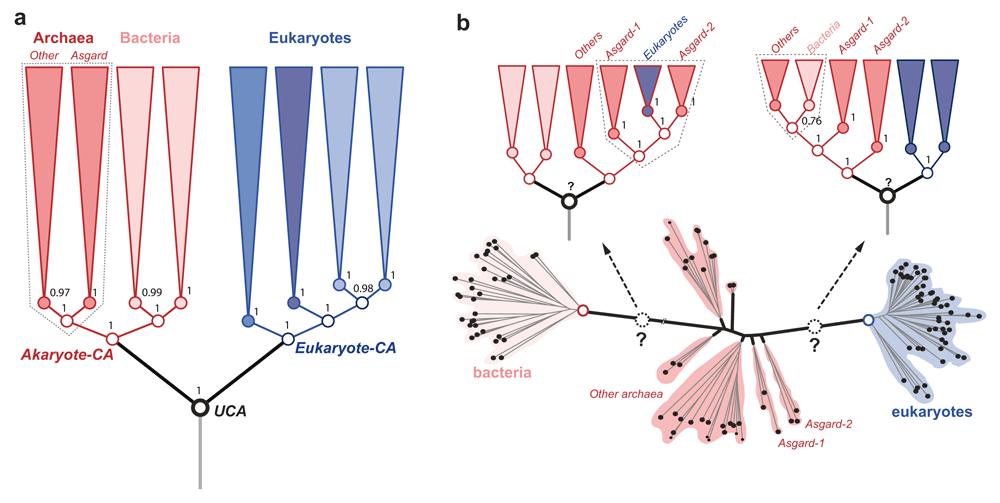
(a) The rooted tree (phylogeny) inferred by estimating the evolution of species-specific changes in protein domain composition. Directional character-evolution models place the root between eukaryotes and akaryotes. Named groups of organisms, including Asgardarchaeota are resolved into clades (i.e. a single ancestor). The Asgard archaea are sister to all other archaea, with euryarchaea being the closest relatives. The phylogeny shown is a condensed form obtained after collapsing the clades of the full tree shown previously1. (b) The unrooted tree inferred by estimating the evolution of protein/gene-specific changes in amino acid composition. The unrooted-tree is the same as in Figure S8d in the article by Williams et al.4. The group archaea, and Asgard archaea are unresolved; and a distinct archaeal ancestor is absent. Time-reversible character evolution models cannot identify the root (the universal common ancestor (UCA)) as well. Alternative rootings polarize the branching order in opposite directions implying incompatible relationships among the major organismal clades. Regardless of the rooting, neither Asgard archaea nor archaea as a whole can be resolved as a monophyletic group. Further, Argards do not share a unique common ancestor with other archaea. Even the best-fitting amino acid evolution models cannot resolve the archaeal radiation despite employing thousands of genes4. The poor resolution of archaea is seen in virtually all trees, with or without inclusion of long branches of bacteria. In such ambiguous cases, “character polarization” as in (a) is likely to be efficient, rather than the more commonly used “graphical polarization” of unrooted trees. Clade support is indicated for key groups as (a) Bayesian posterior probability, (b) bootstrap percentage.
The authors present a comprehensive analysis of protein sequence data and lucid arguments about the fit of the amino acid substitution models to the relevant datasets examined. However, description of the protein domain characters (which they refer to as protein folds) and relevant published analyses were not adequately explained (see references 2 and 3) or were overlooked (see references 1 and 8). Further, based on simple frequency distributions they suspect that identification of UCA and character compositions at the root node could be biased. Such simple frequency distributions can be misleading. Careful and rigorous analyses of empirical datasets1–3,8 that demonstrate the robustness of rooting and tree topology against many potential biases were ignored4. Here, we would like to clarify certain aspects of the published protein domain-based phylogenies so as to avoid further misunderstandings and to highlight their advantages for phylogenetic reconstruction.
Williams et al.4 rely on (i) simulated data to reject a robust phylogeny inferred from empirical data (Figure 1a) that supports the evolutionary kinship of eukaryotes and akaryotes (akaryote 2D-ToL)1–3; and (ii) the so-called bacterial rooting to interpret a partially resolved, unrooted-ToL (Figure 1b), asserting that Asgard archaea are the closest relatives of eukaryotes (eocyte 2D-ToL)4. Both assertions are questionable, since (i) simulated data neither reproduce nor represent empirical distributions, and (ii) poorly resolved trees obscure evolutionary relationships. We argue that Williams et al.4 have overlooked important aspects of assessing phylogenetic signal in empirical data, and that it may be premature to reject a well-supported phylogeny1–3 based on simulated data4.
Reversibility of amino-acid replacements due to biochemical redundancy makes determining character compositions of ancestral nodes ambiguous, as character polarity is ambiguous. This has been a sticking point for locating a distinct archaeal common ancestor (CA) to resolve the archaeal radiation. This is routinely seen as a conspicuous absence of the archaeal CA as well as the universal CA (UCA) in unrooted trees (e.g. Figure 1b), inferred using time-reversible models of character evolution4,9,10. Without a distinct node to unite the archaeal branches, the archaea are unresolved, whereas eukaryotes and bacteria are resolved so that their CA nodes are discernable.
Protein structural domains, unlike amino acids, are biochemically non-redundant (see below) and have proven to be excellent “genomic characters”1,2 that support a robust akaryote 2D-ToL (Figure 1a). Though undervalued, they afford many conceptual and technical advantages over amino acids for reliable phylogenetic modeling1,7,11 and estimating ancestral compositions2,3,12:
Substitutions between structural domains do not occur, unlike amino acid replacements, since each domain defines a distinctive biochemical function1 (Figure 2a).
The natural bias in gain/loss rates, arising from the difficulty of parallel gains and the relative ease of parallel losses, is useful for implementing directional (rooted) character-evolution models3,12,13.
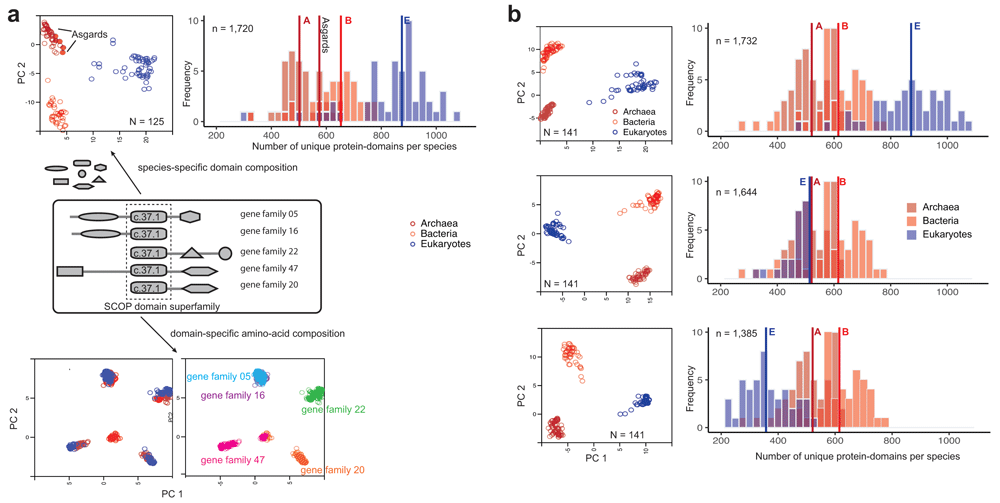
(a) Protein-domains are considered to be independent evolutionary units with a distinct tertiary fold, amino acid sequence and biochemical function. The majority of proteins are multi-domain proteins formed by duplication and recombination of domain units. Covariation of protein-domain composition among the 125 species sampled by Williams et al.4 (top) was compared by principal component analysis (PCA). Each circle in the PCA projection (top left) is a distinct species, defined by a species-specific domain cohort. Asgards are highlighted as filled circles. The frequency distribution (top right) shows the number of distinct protein-domains per species. Vertical intersecting lines in the histograms are the median numbers of protein-domains. Protein domain composition is characteristic of clades of species (top left). In contrast, covariation of amino acid composition (bottom) in a single-domain (super)family is not clade-specific, but gene family-specific. Multiple sequence alignments of a single domain (c.37.1) shared by 5/50 concatenated orthologous gene families from 125 species were sampled for the PCA projection. (b) Effects of severe perturbation of the domain composition in recovering clade-specific distributions was tested in a sample of 141 species. Although it is common to suspect that the rooting between akaryotes and eukaryotes could be biased due to a larger domain cohort in eukaryotes4, it is not the case2,3,12. Diversity of clade-specific domain composition (top right) measured simply as the number of protein domains4 is a poor descriptor of heterogeneity, and can be misleading. Clades are grouped by covarying “protein-domain types”, but not by numbers alone. The rooting is stable and the tree topology is virtually identical even after reducing the eukaryote cohort by 1/3rds (middle) or 2/3rds (bottom)8 of the original composition2. Description of the PCA projections and frequencies are the same as in (a).
A key advantage of non-redundant characters is that estimating ancestral compositions and evolutionary paths of individual characters is much less ambiguous. In addition to identifying the root nodes, an added benefit of the built-in directionality is that mutually exclusive evolutionary fates of individual features – inheritance, loss or transfer – can be resolved efficiently using directional-evolution models1,8,13. Harish et al.1–3 demonstrated that difficult phylogenetic problems can be resolved efficiently by employing protein domain characters and directional evolution models.
To be clear, unrooted trees are not phylogenies per se, since the absence of root-ancestor(s) obscures ancestor-descendant polarity and phylogenetic relatedness14,15. Since identifying the closest relatives of extant groups is the same as determining the closeness of their common ancestors, time-reversible models and unrooted trees remain ineffective tools (Figure 1b). Thus, regardless of the gene-aggregation and tree-reconciliation method used for estimating a consensus unrooted tree4, the location of the archaeal CA or UCA remains ambiguous (Figure 1b). Support from fossils or other sources are not reliable, despite claims to the contrary4. Likewise, predicting the origins of single domains or single genes by estimating amino acid (or nucleotide) compositions also remains ambiguous (reviewed in refs 1,6,7). A sobering revelation is that some datasets/models may be of little use or relevance to resolve questions of deep time evolution – this is sad but true.
Williams et al.4 argue that (i) directional-evolution models12,13 may be unsuitable to predict the unique origin of homologous protein domains; and (ii) the akaryote 2D-ToL1–3 is an unsatisfactory explanation of the evolution of clade-specific compositions of protein domains (Figure 2). Their arguments seem to imply that phylogenetic signal can be recovered only by modeling evolution of amino acid composition. However, the fact that even the best-fitting substitution models are inadequate4, despite ever increasing model complexity to resolve conflicting signals (Figure 1b), suggests that different protein domain-families may require different but incompatible substitution models (Figure 2a). Further, such incompatibilities are likely to make estimating the absolute origins of single-domain families and single genes difficult, since a majority of genes are formed by duplication and recombination of distinct domains1. As a result, distinguishing between gene duplication and horizontal gene transfer, as well as quantifying the extent of duplications and transfers using primary sequences, is highly ambiguous1–3.
The KVR13 model for protein domain data1,2 is an extension of the Markov k states (Mk) model16, a generic probability model for discrete-state characters. A variant at k ≥20 is suitable for modeling evolution of amino acids or copy numbers of gene or protein domain families. While time-reversible variants produce unrooted trees, such directional models consistently recover a 2D phylogeny (Figure 1a) in which akaryotes are the closest relatives of eukaryotes1,2,8. The KVR model assumes that the root ancestor has a different character composition than the rest of the tree, which is essentially an irreversible acyclic process. This is fully consistent with the idea that, on a grand scale, the “tree of life” describes broad generalizations of singular events and major transitions underlying striking sister clade differences. Since parallel evolution of homologous protein-domains or distinct domain permutations is very rare, the KVR model adequately captures the evolution of unique features.
The assumptions of the KVR model are also consistent with the idea that the idiosyncratic compositions of homologous protein-domains (Figure 2) is a characteristic of the clades1–3. In contrast, amino acid compositions in single-domain families are not (Figure 2a). That is, patterns of covariation of species-specific protein-domain compositions clearly distinguish eukaryotes from akaryotes (and archaebacteria from eubacteria). The systematic covariation of homologous domains among the clades is best explained as phylogenetic effect. Consequently, the akaryote 2D-ToL (Figure1a) was consistently recovered with robust support for the major clades regardless of the taxonomic/protein domain diversity sampled (Figure 2b), and regardless of the model complexity1–3,8,12. By contrast, patterns of amino acid covariation are indiscriminate with regard to organismal families, although gene families can be efficiently identified.
The KVR model is an optimal explanation of the evolution of clade-specific composition of homologous features. Complex variants of the KVR model that account for rate variation among both characters and branches also consistently recovered the akaryote 2D-ToL (Figure 1a) despite significantly different model fits1. More complex models are available, such as the no-common-mechanism model17, an extremely parameter-rich model that allows each character to have its own rate, branch length and topology parameters. Even more complex models can be implemented, which assume that the tempo and mode of evolution changes at each internal node along the phylogeny4. However, such over-specified models may not be optimal for generalizing the evolutionary process and may over-fit observed patterns – a form of model misspecification. For instance, empirical datasets are limited to a finite set of homologous protein domains that range between 2,000 and 10,000 characters depending on the protein structure classification scheme1. By contrast, Williams et al.4 use 1,000,000 characters in their simulations to estimate the fit between the simulated data and over-complex models4. That said, it remains to be seen whether more complex models perform better with empirical datasets.
Proteome sequences (predicted protein cohorts from genome sequences) were obtained from recently published studies4,8. Homologous protein structural domains were identified using the homology assignment tools provided by the SUPERFAMILY database as in previous studies1–3. Briefly, each proteome was queried against the hidden Markov model (HMM) library of homologous protein-domains defined at the Superfamily level in the SCOP (Structural Classification of Proteins) hierarchy. The taxonomic diversity of sequenced genomes and the number of unique protein domains identified for each species is shown in Table 1.
Descriptive statistics of protein-domain compositions for each taxonomic sampling, including the frequency distribution and median number of protein domains for each clade (Archaea, Bacteria and Eukarya), were estimated and visualized using the ggplot2 package (v 3.2.1) in R (v3.6.2). Covariation of clade-specific protein-domain composition, as well as domain-specific amino acid composition, was compared using principal component analysis (PCA). Components were generated by an eigenvector decomposition of the character matrix. PCA scores were based on percentage identity of character compositions.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)