Introduction
Healthy human adults produce about 200 billion red blood cells (RBCs) daily to replace those lost by senescence. This process, termed erythropoiesis, is exquisitely regulated by an oxygen-sensing mechanism that has evolved to maintain RBC numbers within a narrow physiological range1–3. Central to this mechanism is erythropoietin (EPO), a cytokine secreted by the kidney in response to low blood oxygen tension. Circulating EPO binds its cognate receptor (EPOR) on bone marrow erythroid progenitors, triggering multiple signaling pathways that support differentiation into mature RBCs. Inherited and acquired abnormalities in EPO production, its downstream activities, or its regulation cause numerous human diseases associated with too many or too few RBCs. The same pathways have been shaped by evolution for adaptation to life under chronic hypoxia at high altitudes. Although much is known about the production of EPO and its biological activities after 40 years of research, the topic remains a rich source for biomedical discovery and therapeutics. This review focuses on recent insights into the oxygen-regulated production of EPO and its actions on post-natal bone marrow erythropoiesis. It is important to note that EPO–EPOR signaling also drives RBC production during embryogenesis through similar but distinct mechanisms4–6.
History of EPO
In 1875, Denis Jourdanet and Paul Bert described anemia-like symptoms in patients living at high altitude and identified low blood oxygen level to be the primary mechanism7. Building on this finding about 30 years later, Carnot and Deflandre discovered that infusion of serum from anemic rabbits into normal ones caused a rise in RBC count, predicting the existence of a circulating factor that stimulates erythropoiesis3. In the early 1950s, studies using parabiotic rats validated the concept of a humoral erythropoiesis-stimulating agent8,9 that was shown by Erslev9 to originate from the kidney10. In 1977, Goldwasser’s group reported the purification of EPO from 2550 liters of urine collected from patients with aplastic anemia11. Molecular cloning of the EPO gene in 1985 facilitated the manufacture of recombinant human EPO (rhEPO) protein for treating various forms of anemia12,13. This work led to discoveries of the EPOR by Lodish’s group in 198914 and subsequently multiple downstream signaling pathways were characterized by many laboratories. An elaborate oxygen-sensing mechanism that regulates EPO production was discovered in the early 1990s by William Kaelin Jr., Sir Peter Ratcliffe, and Gregg Semenza, who received the 2019 Nobel Prize in Physiology or Medicine for this work15–20.
Erythropoietic activities of EPO and EPOR
Multi-potent hematopoietic stem cells undergo a series of differentiation steps that successively restrict developmental potential, giving rise to lineage-committed progenitors (Figure 1)5. The first identifiable erythroid progenitor, termed “burst-forming unit-erythroid” (BFU-E), is defined by its ability to generate large colonies with scattered clusters of erythroblasts in semi-solid medium. Differentiation of BFU-E produces “colony-forming units-erythroid” (CFU-E) that generate smaller colonies containing about 50 cells. Proerythroblasts, the first recognizable erythroid precursor, undergo further maturation steps, which include specialized cell divisions, reduced cell size, elimination of most organelles, development of a specialized cell membrane to facilitate microcirculatory transit, and accumulation of hemoglobin for oxygen transport1,21,22. Terminal erythroid maturation occurs in bone marrow erythroblastic islands composed of erythroid precursors surrounding a central macrophage23. The morphological and functional definitions of committed erythroid progenitors have been augmented by the identification of stage-specific cell surface markers24–31 and, more recently, the discovery of their transcriptional states using single-cell RNA sequencing (scRNAseq)32,33.
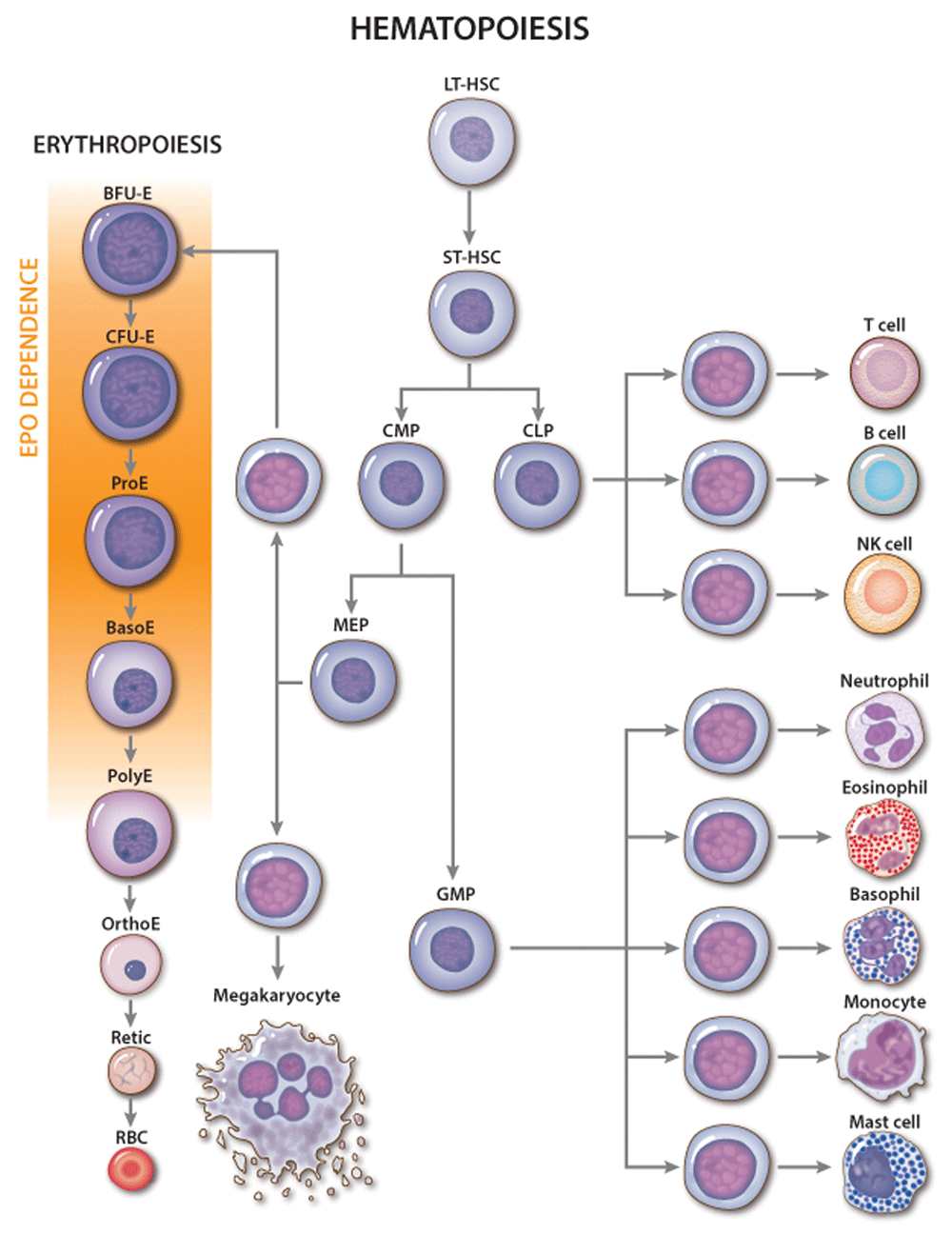
Figure 1. Erythropoietin (EPO) activity during erythropoiesis.
Classic hierarchy of hematopoiesis with stages of red blood cell (RBC) development shown in greater detail. The major site of EPO action is indicated. Genetic and cell culture studies have shown that EPO is required for the development of CFU-E into late-stage erythroblasts. NK, natural killer. Multi-potent hematopoietic progenitors include the following: CLP, common lymphoid progenitor; CMP, common myeloid progenitor; LT-HSC, long-term engrafting hematopoietic stem cell; MEP, megakaryocytic-erythroid progenitor; ST-HSC, short-term hematopoietic stem cell. Committed erythroid progenitors include the following: BFU-E, burst-forming unit-erythroid; CFU-E, colony-forming unit-erythroid. Erythroid precursors include the following: BasoE, basophilic erythroblast; OrthoE, orthochromatic erythroblast; PolyE, polychromatic erythroblast; ProE, proerythroblast; Retic, reticulocyte.
Although multiple cytokines support erythropoiesis34, EPO is the key physiological regulator. Loss of EPO or derangements in EPO signaling in mice or humans cause anemia4,35 while excessive EPO production or EPOR signaling or both cause pathologically increased RBC numbers36–38. EPO acts mainly on CFU-E progenitors and proerythroblasts to maintain their survival and facilitate terminal maturation (Figure 1)25,39–41. Additionally, EPO can stimulate cell proliferation and drive multi-potent hematopoietic progenitors toward an erythroid fate40,42 but is not required for erythroid lineage commitment4. In vivo administration of EPO leads to rapid skewing of multi-potential progenitors away from myeloid and toward the erythroid lineage and to altered gene expression in BFU-E and CFU-E progenitors32.
An oxygen-sensitive feedback loop regulates EPO production
Post-natal EPO production occurs mainly in peritubular fibroblast-like interstitial cells of the kidney43–50 but also in liver, spleen, bone marrow, lungs, and brain51–53 and is regulated by blood oxygen levels through a transcriptional feedback loop (Figure 2)15–19. The hypoxia-inducible transcription factor (HIF) complex binds hypoxia response elements in the EPO gene promoter to stimulate its transcription. Functional HIF is a heterodimer composed of an α subunit (HIFα) and a β subunit (HIFβ, also known as aryl hydrocarbon receptor nuclear translocator or ARNT). The stability of HIF is regulated by prolyl hydroxylase domain (PHD) enzymes, which use oxygen and 2-oxoglutarate to catalyze the hydroxylation of specific proline residues in HIFα, thereby stimulating binding of the HIF heterodimer to the von Hippel–Lindau protein (pVHL) component of an E3 ubiquitin ligase complex3,54,55. Subsequent polyubiquitination of HIF leads to its proteasomal degradation. At low cellular oxygen concentrations, the PHD proteins are inactive and HIF is stabilized for target gene activation. Another 2-oxoglutarate–dependent oxygenase, factor inhibiting HIF (FIH), stimulates the oxygen-dependent hydroxylation of a specific asparagine residue in HIFα, which inhibits its activity by blocking HIFα binding to the transcriptional co-activator p30055,56. In these ways, the PHD and FIH enzymes act as oxygen sensors that inhibit the production of EPO and other HIF targets under oxygen-replete conditions. Remarkably, HIF also activates hundreds of genes besides EPO. Other HIF target genes encode glycolytic enzymes, angiogenic factors, and iron uptake proteins, representing a concerted hypoxia response to increase RBC production, manufacture hemoglobin, enhance tissue perfusion, and promote oxygen-independent metabolism through glycolysis54,57–59.
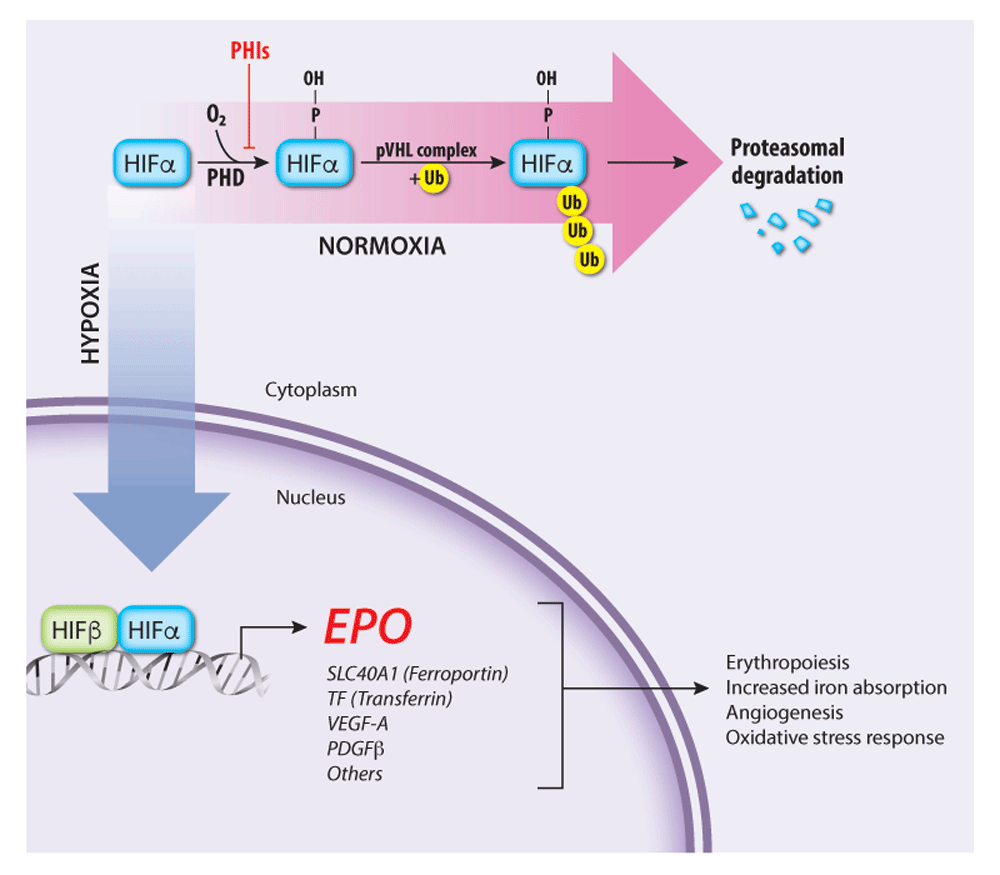
Figure 2. Regulation of endogenous erythropoietin (EPO) gene transcription by the oxygen-sensitive hypoxia-inducible factor (HIF) pathway
The HIF transcription factor heterodimer (HIFα–HIFβ) activates the EPO gene and numerous other genes that promote tissue oxygen delivery. At high oxygen concentrations, prolyl hydroxylase (PHD) enzymes hydroxylate the HIFα subunit, targeting it for ubiquitination by the von Hippel–Lindau protein (pVHL) ubiquitin ligase complex followed by proteasomal degradation. Under hypoxia, PHD enzymes are inactive, thereby stabilizing HIF, which activates transcription of EPO and other target genes involved in tissue oxygen delivery. PHD inhibitors (PHIs) such as roxadustat and vadadustat stabilize HIFα and are under investigation for treating anemia associated with chronic renal failure. PDGFβ, platelet-derived growth factor beta; SLC40A1, solute carrier family 40 member 1; TF, Transferrin; VEGF-A, vascular endothelial growth factor A.
Mammals express three HIFα isoforms (HIF-1α, -2α, and -3α) and three PHD isoforms (PHD1, 2, and 3), each encoded by separate genes with overlapping but distinct tissue distributions and functions60. The production of EPO in adult life is regulated mainly by HIF-2α and PHD261. Perhaps not surprisingly, germline and somatic mutations affecting the PHD–HIF–EPO regulatory pathway are associated with erythrocytosis, anemia, abnormal angiogenesis, and cancer62,63. In mice and humans, loss-of-function mutations in PHD2 and VHL or gain-of-function missense mutations that stabilize HIF-2α by inhibiting its binding to PHD2 or VHL cause erythrocytosis64. An interesting gain-of-function mutation in the EPO gene (c.32delG) was recently identified to cause autosomal dominant erythrocytosis in a multi-generational pedigree65. The single-nucleotide deletion introduces a frameshift into the main EPO mRNA but initiates excess production of EPO from what is normally a non-coding EPO mRNA transcribed from an alternative promoter in intron 1. Variants in the PHD–HIF–EPO pathway have also been selected for in evolution as an adaptive mechanism to living at high altitude. Some of these variants attenuate hypoxia-induced erythrocytosis that can cause deleterious hyperviscosity syndromes64,66–69. These clinical observations highlight the exquisite and complex genetic regulation of EPO production and erythropoiesis. Of note, only one isoform of FIH has been identified. Ablation of the corresponding gene in mice causes metabolic alterations but does not appear to alter the canonical HIF functions in erythropoiesis or angiogenesis70.
EPO activities are mediated through the EPOR
EPO drives erythropoiesis by stimulating the EPOR on the surface of erythroid progenitors. The EPOR is a member of the type I cytokine receptor family distinguished by a conserved extracellular WSXWS amino acid motif, a single-transmembrane domain, and a cytoplasmic tail that lacks intrinsic tyrosine kinase activity71. The proximal cytoplasmic domain of EPOR is bound by the JAK2 tyrosine kinase. Binding of a single EPO molecule to two EPOR molecules triggers a conformational change that stimulates JAK2 to initiate a multi-tiered signaling cascade (Figure 3)72,73. Activated JAK2 phosphorylates itself and several tyrosine residues on the EPOR cytoplasmic tail, which serve as docking sites to engage SH2-containing signaling molecules such as the STAT5 (signal transducer and activator of transcription 5) transcription factor. Following phosphorylation and activation by JAK2, STAT5 enters the nucleus to activate numerous target genes74. Biologically important erythroid STAT5 target genes include the following: BCL2L1, which prevents apoptosis of late-stage erythroblasts75–77; ID1, which promotes erythroblast expansion and survival78; TRIB3, which regulates erythroid maturation79; SPI2A, which encodes a serpin protease with antioxidant activities80; and TFRC (transferrin receptor protein 1), which mediates iron uptake81,82. The recently discovered STAT5 target gene erythroferrone (ERFE) encodes a hormone that acts on hepatocytes to inhibit their production of hepcidin, a different hormone that blocks intestinal iron absorption and release of iron stores from macrophage83. By stimulating the production of ERFE in erythroblasts, EPO increases bioavailable iron for hemoglobin synthesis83,84. In addition to STAT5, EPOR activates the canonical Ras/mitogen-activated protein kinase (MAPK) and phosphoinositide-3 kinase (PI3K)/Akt pathways to enhance erythroid progenitor survival, proliferation, and differentiation3,85–89. The Akt kinase also activates FOXO3, a transcription factor that induces genes that control antioxidant pathways90,91, cell polarity, and enucleation92. Other signaling molecules activated by EPOR include Lyn kinase and PLCγ, although their contributions to erythropoiesis are less clear93,94.
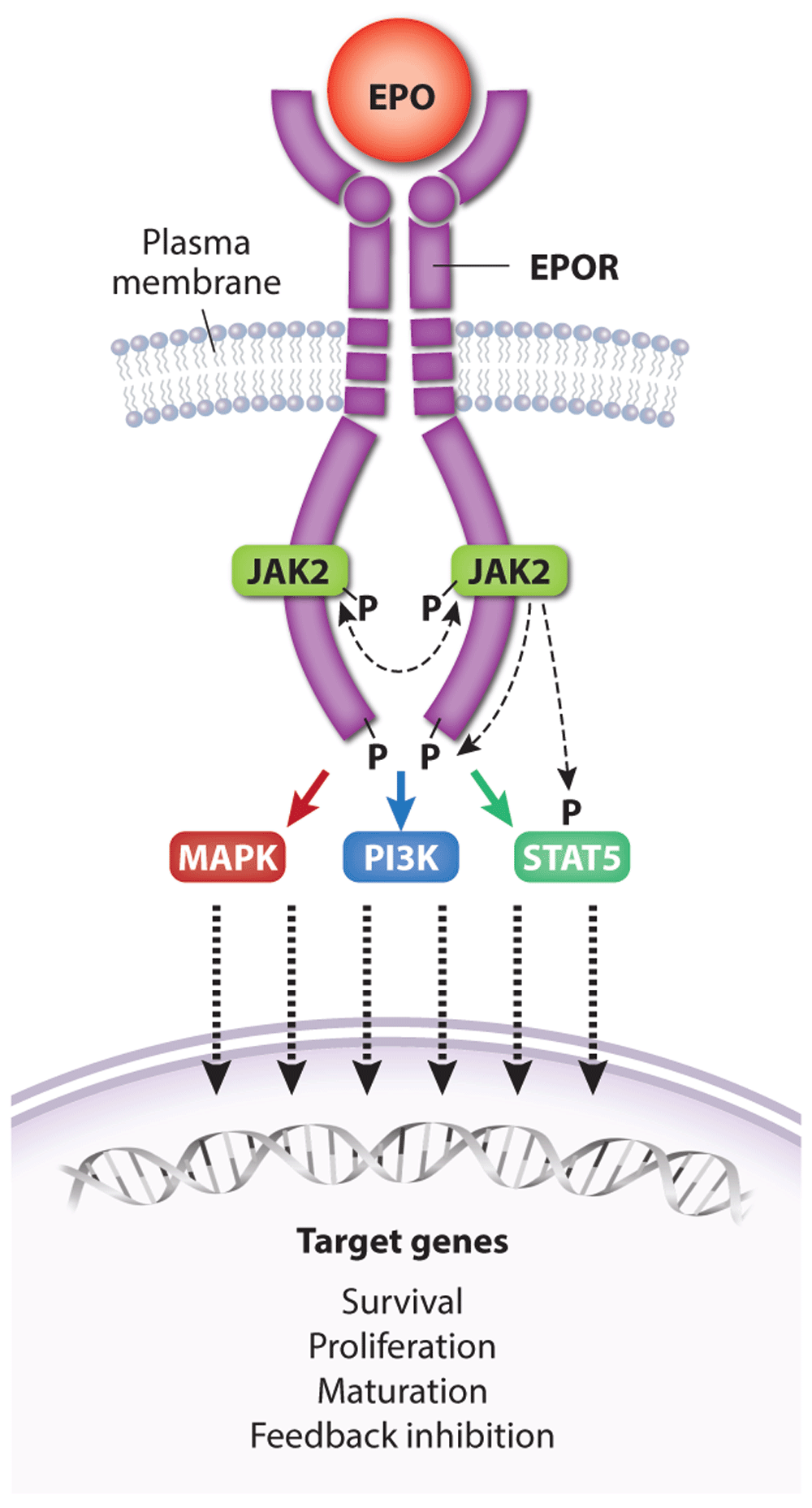
Figure 3. Activation of erythropoietin receptor (EPOR) by EPO.
A single EPO molecule binds and stabilizes EPOR–JAK2 complex dimers, inducing a conformational change that initiates JAK2 trans-phosphorylation and activation. Active JAK2 phosphorylates multiple tyrosine residues on STAT5 and the cytoplasmic domain of EPOR, triggering a signaling cascade that activates numerous effector pathways contributing to biological activity. Dashed lines represent kinase activity. For simplicity, the kinase activity of only one JAK2 protein is indicated. Major signaling pathways activated by EPOR include Ras/MAPK, STAT5, and PI3K/Akt, which drive the expression of genes that promote erythroid progenitor survival, proliferation, and differentiation as well as feedback inhibition of EPOR signaling. MAPK, mitogen-activated protein kinase; PI3K, phosphoinositide-3 kinase; STAT5, signal transducer and activator of transcription 5.
Although some EPO–EPOR effectors can be linked directly to activation of a single linear signaling pathway, overgeneralizing this concept may be biologically inaccurate. As postulated for cytokine receptor signaling in general95, the biological functions of EPOR are likely to be regulated by cross-communications between its numerous downstream signaling pathways and signaling by other cytokine receptors. In regard to the latter, cooperative signaling between the EPOR and stem cell factor receptor (KIT) is believed to promote erythropoiesis96–99. The EPOR also binds the type 2 transferrin receptor (TFR2), which is expressed in hepatocytes and erythroid progenitors. In hepatocytes, the TFR2 stimulates hepcidin production and germline TFR2 mutations cause iron overload (hemochromatosis type 3)100. In erythroid progenitors, TFR2 binds EPOR in the endoplasmic reticulum and facilitates its transport to the cell surface101. The effects of TFR2 in erythroid progenitors appear to be context-dependent and are not fully resolved. In cultured erythroblasts, suppression of TFR2 inhibits erythropoiesis101. In contrast, hematopoietic-specific ablation of the Tfr2 gene in mice enhances erythropoiesis, likely by modulating EPO sensitivity102. Expression of TFR2 in the kidney may inhibit EPO production103. Interaction with iron-bound transferrin stabilizes TFR2 at the cell surface104, representing a potential mechanism by which TFR2 coordinates erythropoietic rate and enteral iron uptake with circulating iron level.
Signaling through EPO–EPOR promotes both basal erythropoiesis, which maintains homeostasis by replacing erythrocytes lost by normal senescence, and “stress erythropoiesis” associated with increased synthetic demands caused by bleeding, excessive RBC destruction, or hypoxia. Relatively low concentrations of EPO during basal erythropoiesis are thought to act mainly by inhibiting apoptosis of erythroid progenitors, while stress erythropoiesis induces higher EPO concentrations that can drive hematopoietic differentiation toward the erythroid fate25,32,40,41. In line with this notion, EPO is able to act like a dimmer switch in activating STAT5 over a wide concentration range105, and functional genomic approaches are beginning to identify direct targets of EPO-activated STAT5 in erythropoiesis74. Different EPO concentrations during basal and stress erythropoiesis are likely to engage distinct signaling modalities, as revealed by a “knock-in” mouse strain in which EPOR is replaced with a truncated version (EPOR-HM) that binds and activates JAK2 but lacks the cytoplasmic portion containing all JAK2 tyrosine substrates106. EPOR-HM mice are viable with a mild defect in steady-state erythropoiesis but are unable to support stress erythropoiesis106. Thus, phosphotyrosine signaling from the EPOR is selectively required for stress erythropoiesis. The signaling pathways that are activated in response to high EPO concentrations differ depending on whether stress is chronic or acute. For example, STAT5-mediated activation of BCL2L1 occurs rapidly after acute bleeding or hypoxia, and then decays, even if high levels of EPO persist. In contrast, persistent or chronic stress conditions such as β-thalassemia elicit a distinct set of EPOR signaling pathways that include the EPOR-mediated suppression of pro-apoptosis genes FAS and BCL2L11 (formerly BIM)32.
EPO signal termination
Activation of EPOR by EPO is balanced by complex negative feedback mechanisms that fine-tune and inhibit signaling to prevent excessive RBC production. Initial evidence for this came from studies of a Finnish family ascertained through an Olympic cross-country skier107. This family and others discovered subsequently were found to have erythrocytosis caused by EPOR-truncating mutations that eliminate portions of the cytoplasmic domain, which later was found to negatively regulate EPOR signaling by recruiting various inhibitory proteins, including the tyrosine phosphatase PTPN6, members of the suppressor of cytokine signaling (SOCS) protein family, SH2B adapter protein 3 (SH2B3, LNK), and the p85 regulatory subunit of PI3K38. Mechanistically, PTPN6 attenuates EPOR signaling by dephosphorylating JAK2108,109. CISH and SOCS3 block access of STAT5 to the EPOR, whereas SOCS1 binds to the JAK2 kinase domain and reduces its tyrosine kinase activity110. Transcription of SOCS1, SOCS3, and CISH are induced by STAT5, forming a negative feedback loop111. Mutations in JAK2 at the SOCS3 binding site and mutations in SOCS3 occur in patients with erythrocytosis112,113. The SH2B3 protein (LNK) is upregulated and phosphorylated in response to EPO and inhibits EPOR signaling by binding phosphotyrosine residues in JAK2 and the cytoplasmic tail of EPOR114,115. Sh2b3−/− mice exhibit features of myeloproliferative neoplasms (MPNs) such as splenomegaly and extramedullary hematopoiesis, and inactivating SH2B3 mutations are associated with myeloproliferative disease in humans115. Genome-wide association studies have identified a hypomorphic SH2B3 variant associated with elevated hemoglobin and RBC counts116,117, and suppression of SH2B3 production by RNA interference improved the production of RBCs by in vitro differentiation of human CD34+ cells and embryonic stem cells118.
The EPOR is also negatively regulated at the protein level by several mechanisms119,120. First, the p85 protein, which facilitates EPOR signaling as a regulatory subunit for PI3K121, also promotes EPOR endocytosis and degradation122,123. Upon EPO stimulation, the casitas B-lineage lymphoma (CBL) protein ubiquitinates p85 bound to the cytoplasmic domain of EPOR, facilitating interaction with the adaptor protein Epsin-1 to promote endocytosis. Second, prolyl hydroxylase D3 (PHD3)-mediated proline hydroxylation of EPOR stimulates its proteasomal degradation124. Third, iron deficiency reduces the expression of EPOR through interactions with TFR2 and Scribble, a scaffold protein that facilitates EPOR recycling125. This mechanism may explain EPO resistance associated with iron deficiency. Dipeptidylpeptidase (DPP4, CD26) expressed on hematopoietic and stromal cells truncates EPO into inactive fragments, reducing its plasma activity126. These examples illustrate how EPOR signaling is terminated by many proteins acting through multiple mechanisms, most of which are components of a negative feedback loop triggered by EPOR activation.
Recent insights into EPO–EPOR signaling
The discovery of activating JAK2 mutations in MPNs has fueled the development of ruxolitinib and other JAK2 inhibitors. Ruxolitinib induces clinical responses and improves survival in some patients with MPN but its overall effects and therapeutic index are relatively modest127. New structure–function studies of EPOR and JAK2 may inform the rational design of novel drugs for MPNs. Binding of JAK2 to nascent EPOR in the endoplasmic reticulum facilitates its trafficking to the plasma membrane128. The importance of this protein interaction is freshly reinforced by findings that MPN-associated JAK2 mutants use EPOR as a scaffold for recruiting downstream substrates in order to drive EPO-independent erythrocytosis129. Moreover, changes in the JAK2 pseudokinase domain, which does not interact with EPOR directly, can affect EPOR–JAK2 association (Figure 4)129. Single-molecule fluorescence microscopy showed that EPO stimulates self-association of EPOR-bound JAK2 through its pseudokinase domain and that MPN-associated JAK2 mutations strengthen this interaction in the absence of EPO130. Thus, mutant JAK2 proteins drive EPO-independent EPOR signaling by enhancing dimerization of EPOR–JAK2 complexes. Mutant JAK2 also drives MPN by stimulating ligand-independent activation of the thrombopoietin receptor, which is structurally similar to the EPOR. The authors note that MPNs might be treated by drugs which inhibit self-interaction of the JAK2 pseudokinase domain. Although JAK2 binds EPOR through its cytoplasmic box 1, subsequent activation requires another EPOR conserved region, termed the “hydrophobic switch”131. Crystallographic data suggest that this region positions EPOR–JAK2 molecules into a specific conformation that facilitates JAK2 activation132.
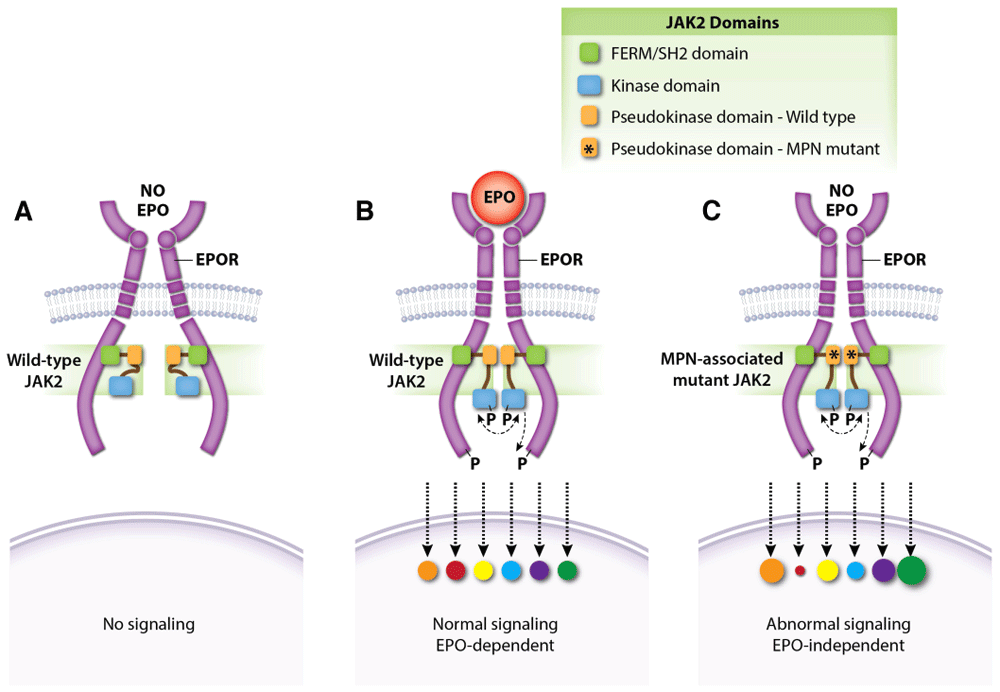
Figure 4. JAK2 regulates dimerization of the EPOR–JAK2 complex.
(A) Normal EPOR–JAK2 complexes are inert without EPO. (B) EPO binding to EPOR stabilizes the EPOR–JAK2 complex and triggers downstream signaling by activating JAK2. Colored circles represent normalized activation levels of EPOR signaling targets. (C) Myeloproliferative neoplasm (MPN)-associated mutations in the JAK2 pseudokinase domain, which does not interact directly with EPOR, stabilize dimerization of EPOR–JAK2 complexes and activate JAK2 in the absence of EPO. Mutations in the linker region separating the FERM-SH2 and pseudokinase domains (exon 12) act similarly (not shown). Constitutive activation of the EPOR by MPN-associated mutations causes abnormal downstream signaling relative to that induced by EPO142–146. EPO, erythropoietin; EPOR, erythropoietin receptor.
Activated EPOR triggers multiple signaling pathways that interact to specify the activation of different effectors and biological output. Medically relevant insights into this problem were gained by the discovery of a patient with pure red cell aplasia (Diamond–Blackfan anemia) caused by a homozygous EPO gene missense mutation (R150Q)133. Although the mutant EPO protein exhibited only threefold reduced steady-state affinity for EPOR, kinetic studies revealed faster on-rate (kon) and off-rate (koff) (Figure 5). Abnormally rapid release of the mutant EPO from EPOR was associated with impaired EPOR dimerization and reduced JAK2 activation. Remarkably, alterations in downstream phospho-signaling elicited by the mutant EPO were highly selective. Thus, erythropoietic failure was not caused by complete loss of EPO activity but rather by altered function. This study shows how variability in ligand-induced conformational changes of a cytokine receptor (in this case, EPO on- and off-rates) can selectively alter downstream signaling and biology.
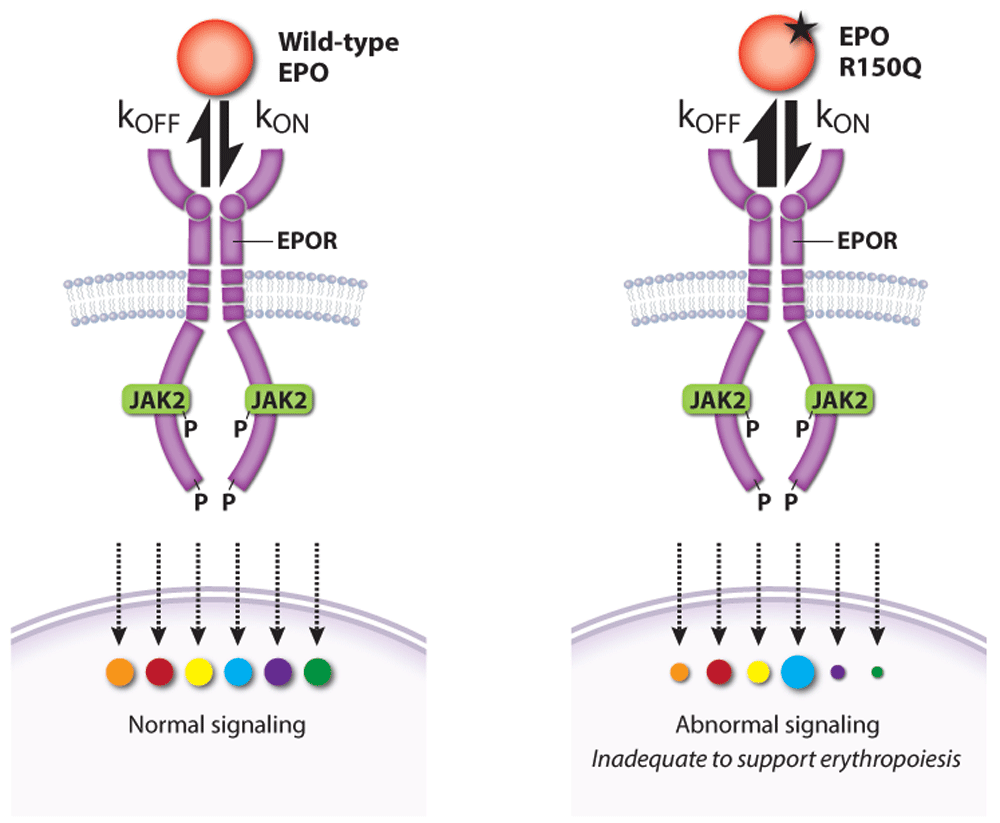
Figure 5. A pathological erythropoietin (EPO) mutant with altered binding kinetics to EPO receptor (EPOR) causes qualitative changes in downstream signaling
A homozygous p.R150Q EPO mutation was discovered in a patient with severe anemia caused by pure red cell aplasia (Diamond–Blackfan anemia). High levels of the mutant EPO failed to restore erythropoiesis despite an only threefold reduction in its overall affinity for EPOR. Compared with wild-type EPO, the mutant EPO interaction with EPOR was kinetically biased with higher on- and off-rates that altered the activation of specific EPOR effector pathways. kon, rate of association; koff, rate of dissociation. This figure was created using data from Kim et al. 2017133.
Two other studies examined EPO–EPOR structure–function relationships more systematically by designing a series of EPOR ligands that generate different homodimer topologies, resulting in qualitative variation in signaling output73,134. These findings have potential medical implications. For example, one study showed that different artificial ligands that resulted in different angles and distance between EPOR homodimer subunits generated unique signaling patterns with stage-selective effects on hematopoiesis (Figure 6)134. The other study73 identified artificial EPOR ligands that can block EPO-independent signaling by the MPN-associated mutation JAK2V617F, which may inform new therapies for MPNs134. Overall, understanding and controlling the signaling output of different EPOR–JAK2 homo-dimer conformations may be used to precisely manipulate hematopoiesis or suppress pathologically active signaling. In this regard, many such studies performed on EPOR are generalizable to other cytokine receptors135–137.
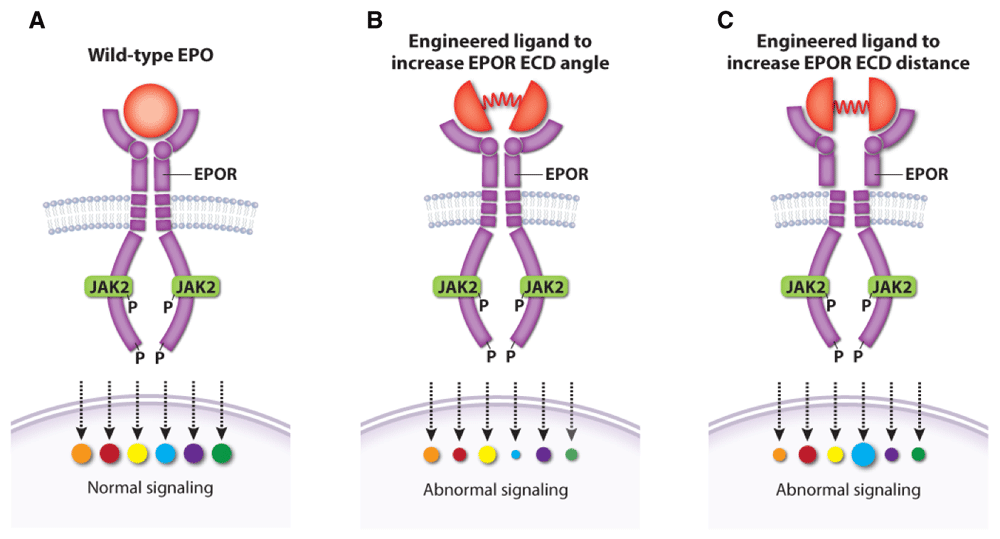
Figure 6. Altered topology of the erythropoietin receptor (EPOR) extracellular domains (ECDs) induced by engineered ligands produces qualitative changes in downstream signaling.
(A) Wild-type erythropoietin (EPO) causes normal activation of EPOR signaling targets. Engineered EPOR ligands that modify the angle (B) or distance (C) between EPOR ECDs produce selective alterations in the activation of downstream signaling targets. This figure was created using data from Mohan et al., 2019134.
Pharmacologic stimulation of erythropoiesis
rhEPO is used to treat anemia associated with a variety of diseases13,138. The most common indication is anemia of chronic renal failure where rhEPO increases blood hemoglobin levels and improves quality of life139. In general, patients with renal failure have high levels of hepcidin, which limits iron absorption and availability for erythropoiesis140,141. Thus, rhEPO therapy usually requires administration of intravenous iron139. Although the benefits of rhEPO in renal failure are clear, its overaggressive use is associated with increased rates of arteriovenous fistula thrombosis, venous thromboembolism, congestive heart failure, myocardial infarction, and death138,147–150. Similarly, rhEPO use for cancer-related anemia has been associated with reduced survival151. Elevated blood viscosity caused by increased RBC mass probably contributes to these adverse events. Additionally, some adverse effects of rhEPO may result from stimulation of EPOR signaling in non-erythroid tissues or tumor cells or both151–157, although this point is complicated by technical difficulties in establishing the presence of EPOR in non-erythroid tissues because of non-specific antibody interactions158. Regardless, current guidelines recommend careful titration of rhEPO dosing in patients with renal failure139. A recent study showed that, compared with low-dose intravenous iron sucrose, high-dose intravenous iron sucrose therapy in chronic renal failure resulted in reduced dosage requirements for rhEPO, fewer major adverse cardiovascular events, and lower death rates159.
The routine use of rhEPO in most cancer patients who are undergoing curative chemotherapy should be avoided160. rhEPO remains an important drug for treating anemia associated with myelodysplastic syndrome, although responses are often transient161–163. From a historical perspective, the first rhEPO (epoetin alfa) was approved for clinical use in 1989 and the longer-acting darbepoetin was approved in 2001. Combined sales reached about $5 billion per year in 2005 and then declined by 40% over the next 6 years as the price of the drugs dropped and the potential adverse effects became recognized164.
The prolyl hydroxylase inhibitors (PHIs), which act by stabilizing HIFα to stimulate endogenous EPO production (Figure 2), are promising new agents for treating anemia of chronic kidney disease and perhaps other etiologies165,166. Numerous clinical studies have shown that PHIs are effective for raising hemoglobin levels in subjects with chronic renal failure166–170. Compared with rhEPO, PHIs offer several potential advantages, including oral administration, improved iron utilization possibly due to suppression of hepcidin, lowering of plasma lipids and cholesterol, and efficacy at relatively low plasma concentrations of endogenous EPO, which may reduce cardiovascular toxicities. Three PHIs are in advanced phase III clinical development, and one was recently approved for clinical use in China171. Although the drugs have been shown to be relatively safe in clinical trials, there are numerous theoretical concerns related to on-target effects given the extensive number of genes and biological pathways that are regulated by the HIF transcription factors. Potential adverse effects include alterations in metabolism, immune response, vascular tone, and angiogenesis. Monitoring for these problems is required in more extended clinical trials pre- and post-marketing.
Conclusions
EPO and its receptor are essential for the differentiation of CFU-E progenitors into mature RBCs. The complex, multi-layered biochemical pathways that regulate EPO production, signal through EPO engagement of EPOR, and extinguish EPOR signaling are all geared to maintain circulating RBC numbers in a narrow physiological range at steady state and during erythropoietic stress. Examination of these processes over more than 40 years has elucidated fundamental concepts of general biology, defined the mechanisms of human diseases associated with over- or under-production of RBCs, and produced a remarkably useful biological drug to treat some forms of anemia. The development of rhEPO, and more recently PHD inhibitors, arose from basic biological research and represents excellent paradigms for “bench to bedside and back” therapeutic development. Despite the tremendous knowledge gained through extensive studies of EPO and EPOR over many years, the field remains a fruitful area of research, as illustrated by ongoing efforts to better understand the complexities of the PHD–HIF–EPO pathway, structure–function regulation of EPO–EPOR–JAK2 signaling, and mechanisms of human disease caused by germline and somatic alterations in genes tied to EPO biology. In fact, interesting and medically relevant research problems related to EPO are too numerous to cover in a single review. Topics not covered here include the biology of EPOR signaling in non-erythroid tissues and its role in metabolic pathways149,151,172–179. Thus, it is likely that laboratory scientists and clinical researchers who study EPO-related biology and medicine will continue to generate exciting and clinically useful findings for many years to come.
Acknowledgments
We thank Anita Impagliazzo, CMI (Anita Impagliazzo Medical Illustration) for assistance with the figures and Merav Socolovsky (Department of Molecular, Cell and Cancer Biology and Department of Pediatric, Division of Hematology/Oncology at the University of Massachusetts Medical School) for critical review of the manuscript.
Faculty Opinions recommendedReferences
- 1.
Moras M, Lefevre SD, Ostuni MA:
From Erythroblasts to Mature Red Blood Cells: Organelle Clearance in Mammals.
Front Physiol.
2017; 8: 1076. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 2.
Koury MJ:
Abnormal erythropoiesis and the pathophysiology of chronic anemia.
Blood Rev.
2014; 28(2): 49–66. PubMed Abstract
| Publisher Full Text
- 3.
Bunn HF:
Erythropoietin.
Cold Spring Harb Perspect Med.
2013; 3(3): a011619. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 4.
Wu H, Liu X, Jaenisch R, et al.:
Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor.
Cell.
1995; 83(1): 59–67. PubMed Abstract
| Publisher Full Text
- 5.
Palis J:
Primitive and definitive erythropoiesis in mammals.
Front Physiol.
2014; 5: 3. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 6.
Malik J, Kim AR, Tyre KA, et al.:
Erythropoietin critically regulates the terminal maturation of murine and human primitive erythroblasts.
Haematologica.
2013; 98(11): 1778–87. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 7.
West JB, Richalet JP:
Denis Jourdanet (1815-1892) and the early recognition of the role of hypoxia at high altitude.
Am J Physiol Lung Cell Mol Physiol.
2013; 305(5): L333–40. PubMed Abstract
| Publisher Full Text
- 8.
Reissmann KR:
Studies on the mechanism of erythropoietic stimulation in parabiotic rats during hypoxia.
Blood.
1950; 5(4): 372–80. PubMed Abstract
- 9.
Erslev A:
Humoral Regulation of Red Cell Production.
Blood.
1953; 8(4): 349–57. PubMed Abstract
- 10.
Jacobson LO, Goldwasser E, Fried W, et al.:
Role of the kidney in erythropoiesis.
Nature.
1957; 179: 633–4. Publisher Full Text
- 11.
Miyake T, Kung CK, Goldwasser E:
Purification of human erythropoietin.
J Biol Chem.
1977; 252(15): 5558–64. PubMed Abstract
- 12.
Lin FK, Suggs S, Lin CH, et al.:
Cloning and expression of the human erythropoietin gene.
Proc Natl Acad Sci U S A.
1985; 82(22): 7580–4. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 13.
Kalantar-Zadeh K:
History of Erythropoiesis-Stimulating Agents, the Development of Biosimilars, and the Future of Anemia Treatment in Nephrology.
Am J Nephrol.
2017; 45(3): 235–47. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 14.
D’Andrea AD, Lodish HF, Wong GG:
Expression cloning of the murine erythropoietin receptor.
Cell.
1989; 57(2): 277–85. PubMed Abstract
| Publisher Full Text
- 15.
Semenza GL, Nejfelt MK, Chi SM, et al.:
Hypoxia-inducible nuclear factors bind to an enhancer element located 3' to the human erythropoietin gene.
Proc Natl Acad Sci U S A.
1991; 88(13): 5680–4. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 16.
Maxwell PH, Wiesener MS, Chang GW, et al.:
The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis.
Nature.
1999; 399(6733): 271–5. PubMed Abstract
| Publisher Full Text
- 17.
Wang GL, Jiang BH, Rue EA, et al.:
Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension.
Proc Natl Acad Sci U S A.
1995; 92(12): 5510–4. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 18.
Ivan M, Kondo K, Yang H, et al.:
HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing.
Science.
2001; 292(5516): 464–8. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 19.
Jaakkola P, Mole DR, Tian YM, et al.:
Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation.
Science.
2001; 292(5516): 468–72. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 20.
Johnson R:
How cells sense and adapt to oxygen availability. Stockholm: Nobel Foundation; 2019 [cited 2020 June]. Reference Source
- 21.
Dzierzak E, Philipsen S:
Erythropoiesis: Development and differentiation.
Cold Spring Harb Perspect Med.
2013; 3(4): a011601. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 22.
Jelkmann W:
Functional significance of erythrocytes. In: Lang F, Foller M, editors. Erythrocytes. London: Imperial College Press; 2012. 1–56. Publisher Full Text
- 23.
Lee SH, Crocker PR, Westaby S, et al.:
Isolation and immunocytochemical characterization of human bone marrow stromal macrophages in hemopoietic clusters.
J Exp Med.
1988; 168(3): 1193–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 24.
Chen K, Liu J, Heck S, et al.:
Resolving the distinct stages in erythroid differentiation based on dynamic changes in membrane protein expression during erythropoiesis.
Proc Natl Acad Sci U S A.
2009; 106(41): 17413–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 25.
Liu Y, Pop R, Sadegh C, et al.:
Suppression of Fas-FasL coexpression by erythropoietin mediates erythroblast expansion during the erythropoietic stress response in vivo.
Blood.
2006; 108(1): 123–33. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 26.
Pronk CJH, Rossi DJ, Månsson R, et al.:
Elucidation of the phenotypic, functional, and molecular topography of a myeloerythroid progenitor cell hierarchy.
Cell Stem Cell.
2007; 1(4): 428–42. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 27.
Hu J, Liu J, Xue F, et al.:
Isolation and functional characterization of human erythroblasts at distinct stages: Implications for understanding of normal and disordered erythropoiesis in vivo.
Blood.
2013; 121(16): 3246–53. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 28.
Ludwig LS, Lareau CA, Bao EL, et al.:
Transcriptional States and Chromatin Accessibility Underlying Human Erythropoiesis.
Cell Rep.
2019; 27(11): 3228–3240.e7. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 29.
Li J, Hale J, Bhagia P, et al.:
Isolation and transcriptome analyses of human erythroid progenitors: BFU-E and CFU-E.
Blood.
2014; 124(24): 3636–45. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 30.
An X, Schulz VP, Li J, et al.:
Global transcriptome analyses of human and murine terminal erythroid differentiation.
Blood.
2014; 123(22): 3466–77. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 31.
Schulz VP, Yan H, Lezon-Geyda K, et al.:
A Unique Epigenomic Landscape Defines Human Erythropoiesis.
Cell Rep.
2019; 28(11): 2996–3009.e7. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 32.
Tusi BK, Wolock SL, Weinreb C, et al.:
Population snapshots predict early haematopoietic and erythroid hierarchies.
Nature.
2018; 555(7694): 54–60. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 33.
Pellin D, Loperfido M, Baricordi C, et al.:
A comprehensive single cell transcriptional landscape of human hematopoietic progenitors.
Nat Commun.
2019; 10(1): 2395. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 34.
Lodish H, Flygare J, Chou S:
From stem cell to erythroblast: Regulation of red cell production at multiple levels by multiple hormones.
IUBMB Life.
2010; 62(7): 492–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 35.
Parganas E, Wang D, Stravopodis D, et al.:
Jak2 Is Essential for Signaling through a Variety of Cytokine Receptors.
Cell.
1998; 93(3): 385–95. PubMed Abstract
| Publisher Full Text
- 36.
Hammond D, Winnick S:
Paraneoplastic erythrocytosis and ectopic erythropoietins.
Ann N Y Acad Sci.
1974; 230: 219–27. PubMed Abstract
| Publisher Full Text
- 37.
Gross M, Ben-Califa N, McMullin MF, et al.:
Polycythaemia-inducing mutations in the erythropoietin receptor (EPOR): Mechanism and function as elucidated by epidermal growth factor receptor-EPOR chimeras.
Br J Haematol.
2014; 165(4): 519–28. PubMed Abstract
| Publisher Full Text
- 38.
Huang LJ, Shen YM, Bulut GB:
Advances in understanding the pathogenesis of primary familial and congenital polycythaemia.
Br J Haematol.
2010; 148(6): 844–52. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 39.
Koury M, Bondurant M:
Erythropoietin retards DNA breakdown and prevents programmed death in erythroid progenitor cells.
Science.
1990; 248(4953): 378–81. PubMed Abstract
| Publisher Full Text
- 40.
Spivak JL, Pham T, Isaacs M, et al.:
Erythropoietin is both a mitogen and a survival factor.
Blood.
1991; 77(6): 1228–33. PubMed Abstract
- 41.
Koury MJ, Bondurant MC, Atkinson JB:
Erythropoietin control of terminal erythroid differentiation: maintenance of cell viability, production of hemoglobin, and development of the erythrocyte membrane.
Blood Cells.
1987; 13(1–2): 217–26. PubMed Abstract
- 42.
Grover A, Mancini E, Moore S, et al.:
Erythropoietin guides multipotent hematopoietic progenitor cells toward an erythroid fate.
J Exp Med.
2014; 211(2): 181–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 43.
Bachmann S, Le Hir M, Eckardt KU:
Co-localization of erythropoietin mRNA and ecto-5'-nucleotidase immunoreactivity in peritubular cells of rat renal cortex indicates that fibroblasts produce erythropoietin.
J Histochem Cytochem.
1993; 41(3): 335–41. PubMed Abstract
| Publisher Full Text
- 44.
Lacombe C, Da Silva JL, Bruneval P, et al.:
Peritubular cells are the site of erythropoietin synthesis in the murine hypoxic kidney.
J Clin Invest.
1988; 81(2): 620–3. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 45.
Koury ST, Bondurant MC, Koury MJ:
Localization of erythropoietin synthesizing cells in murine kidneys by in situ hybridization.
Blood.
1988; 71(2): 524–7. PubMed Abstract
- 46.
Zeisberg M, Kalluri R:
Physiology of the Renal Interstitium.
Clin J Am Soc Nephrol.
2015; 10(10): 1831–40. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 47.
Fisher JW, Koury S, Ducey T, et al.:
Erythropoietin production by interstitial cells of hypoxic monkey kidneys.
Br J Haematol.
1996; 95(1): 27–32. PubMed Abstract
| Publisher Full Text
- 48.
Shanks JH, Hill CM, Lappin TR, et al.:
Localization of erythropoietin gene expression in proximal renal tubular cells detected by digoxigenin-labelled oligonucleotide probes.
J Pathol.
1996; 179(3): 283–7. PubMed Abstract
| Publisher Full Text
- 49.
Pan X, Suzuki N, Hirano I, et al.:
Isolation and characterization of renal erythropoietin-producing cells from genetically produced anemia mice.
PLoS One.
2011; 6(10): e25839. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 50.
Kurtz A:
Endocrine functions of the renal interstitium.
Pflugers Arch.
2017; 469(7–8): 869–76. PubMed Abstract
| Publisher Full Text
- 51.
Lacombe C, Da Silva JL, Bruneval P, et al.:
Erythropoietin: Sites of synthesis and regulation of secretion.
Am J Kidney Dis.
1991; 18(4 Suppl 1): 14–9. PubMed Abstract
- 52.
Jelkmann W:
Regulation of erythropoietin production.
J Physiol.
2011; 589(Pt 6): 1251–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 53.
Maxwell PH, Ferguson DJ, Nicholls LG, et al.:
Sites of erythropoietin production.
Kidney Int.
1997; 51(2): 393–401. PubMed Abstract
| Publisher Full Text
- 54.
Haase VH:
Regulation of erythropoiesis by hypoxia-inducible factors.
Blood Rev.
2013; 27(1): 41–53. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 55.
Schofield CJ, Ratcliffe PJ:
Oxygen sensing by HIF hydroxylases.
Nat Rev Mol Cell Biol.
2004; 5(5): 343–54. PubMed Abstract
| Publisher Full Text
- 56.
Mahon PC, Hirota K, Semenza GL:
FIH-1: A novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity.
Genes Dev.
2001; 15(20): 2675–86. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 57.
Peyssonnaux C, Nizet V, Johnson RS:
Role of the hypoxia inducible factors HIF in iron metabolism.
Cell Cycle.
2008; 7(1): 28–32. PubMed Abstract
| Publisher Full Text
- 58.
Masoud GN, Li W:
HIF-1α pathway: Role, regulation and intervention for cancer therapy.
Acta Pharm Sin B.
2015; 5(5): 378–89. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 59.
Ratcliffe PJ:
HIF-1 and HIF-2: Working alone or together in hypoxia?
J Clin Invest.
2007; 117(4): 862–5. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 60.
Kaplan JM, Sharma N, Dikdan S:
Hypoxia-Inducible Factor and Its Role in the Management of Anemia in Chronic Kidney Disease.
Int J Mol Sci.
2018; 19(2): 389. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 61.
Appelhoff RJ, Tian YM, Raval RR, et al.:
Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor.
J Biol Chem.
2004; 279(37): 38458–65. PubMed Abstract
| Publisher Full Text
- 62.
Simonson TS, Yang Y, Huff CD, et al.:
Genetic Evidence for High-Altitude Adaptation in Tibet.
Science.
2010; 329(5987): 72–5. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 63.
Chappell JC, Payne LB, Rathmell WK:
Hypoxia, angiogenesis, and metabolism in the hereditary kidney cancers.
J Clin Invest.
2019; 129(2): 442–51. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 64.
Lappin TR, Lee FS:
Update on mutations in the HIF: EPO pathway and their role in erythrocytosis.
Blood Rev.
2019; 37: 100590. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 65.
Zmajkovic J, Lundberg P, Nienhold R, et al.:
A Gain-of-Function Mutation in EPO in Familial Erythrocytosis.
N Engl J Med.
2018; 378(10): 924–30. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 66.
Bigham AW:
Genetics of human origin and evolution: High-altitude adaptations.
Curr Opin Genet Dev.
2016; 41: 8–13. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 67.
Julian CG, Moore LG:
Human Genetic Adaptation to High Altitude: Evidence from the Andes.
Genes (Basel).
2019; 10(2): 150. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 68.
van Tissot Patot MC, Gassmann M:
Hypoxia: Adapting to High Altitude by Mutating EPAS-1, the Gene Encoding HIF-2α.
High Alt Med Biol.
2011; 12(2): 157–67. PubMed Abstract
| Publisher Full Text
- 69.
Yi X, Liang Y, Huerta-Sanchez E, et al.:
Sequencing of 50 human exomes reveals adaptation to high altitude.
Science.
2010; 329(5987): 75–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 70.
Zhang N, Fu Z, Linke S, et al.:
The asparaginyl hydroxylase factor inhibiting HIF-1alpha is an essential regulator of metabolism.
Cell Metab.
2010; 11(5): 364–78. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 71.
Constantinescu SN:
Mechanism of erythropoietin receptor activation. In: Elliott S, Foote M, Molineux G editors. Erythropoietins, Erythropoietic Factors, and Erythropoiesis. Basel: Birkhäuser Basel; 2009; 175–98. Reference Source
- 72.
Constantinescu SN, Keren T, Socolovsky M, et al.:
Ligand-independent oligomerization of cell-surface erythropoietin receptor is mediated by the transmembrane domain.
Proc Natl Acad Sci U S A.
2001; 98(8): 4379–84. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 73.
Moraga I, Wernig G, Wilmes S, et al.:
Tuning cytokine receptor signaling by re-orienting dimer geometry with surrogate ligands.
Cell.
2015; 160(6): 1196–208. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 74.
Gillinder KR, Tuckey H, Bell CC, et al.:
Direct targets of pSTAT5 signalling in erythropoiesis.
PLoS One.
2017; 12(7): e0180922. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 75.
Rhodes MM, Kopsombut P, Bondurant MC, et al.:
Bcl-xL prevents apoptosis of late-stage erythroblasts but does not mediate the antiapoptotic effect of erythropoietin.
Blood.
2005; 106(5): 1857–63. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 76.
Socolovsky M, Fallon AEJ, Wang S, et al.:
Fetal anemia and apoptosis of red cell progenitors in Stat5a-/-5b-/- mice: a direct role for Stat5 in Bcl-X(L) induction.
Cell.
1999; 98(2): 181–91. PubMed Abstract
| Publisher Full Text
- 77.
Cui Y, Riedlinger G, Miyoshi K, et al.:
Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation.
Mol Cell Biol.
2004; 24(18): 8037–47. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 78.
Wood AD, Chen E, Donaldson IJ, et al.:
ID1 promotes expansion and survival of primary erythroid cells and is a target of JAK2V617F-STAT5 signaling.
Blood.
2009; 114(9): 1820–30. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 79.
Dev A, Asch R, Jachimowicz E, et al.:
Governing roles for Trib3 pseudokinase during stress erythropoiesis.
Exp Hematol.
2017; 49: 48–55.e5. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 80.
Dev A, Byrne SM, Verma R, et al.:
Erythropoietin-directed erythropoiesis depends on serpin inhibition of erythroblast lysosomal cathepsins.
J Exp Med.
2013; 210(2): 225–32. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 81.
Zhu BM, McLaughlin SK, Na R, et al.:
Hematopoietic-specific Stat5-null mice display microcytic hypochromic anemia associated with reduced transferrin receptor gene expression.
Blood.
2008; 112(5): 2071–80. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 82.
Kerenyi MA, Grebien F, Gehart H, et al.:
Stat5 regulates cellular iron uptake of erythroid cells via IRP-2 and TfR-1.
Blood.
2008; 112(9): 3878–88. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 83.
Kautz L, Jung G, Valore EV, et al.:
Identification of erythroferrone as an erythroid regulator of iron metabolism.
Nat Genet.
2014; 46(7): 678–84. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 84.
Coffey R, Ganz T:
Erythroferrone: an erythroid regulator of hepcidin and iron metabolism.
Hemasphere.
2018; 2(2): e35. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 85.
Rainville N, Jachimowicz E, Wojchowski DM:
Targeting EPO and EPO receptor pathways in anemia and dysregulated erythropoiesis.
Expert Opin Ther Targets.
2016; 20(3): 287–301. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 86.
Wojchowski DM, Sathyanarayana P, Dev A:
Erythropoietin receptor response circuits.
Curr Opin Hematol.
2010: 17(3): 169–76. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 87.
Lodish HF, Ghaffari S, Socolovsky M, et al.:
Intracellular signaling by the erythropoietin receptor. In: Elliott S, Foote M, Molineux G, editors. Erythropoietins, Erythropoietic Factors, and Erythropoiesis. Basel: Birkhäuser Basel; 2009; 155–74. Publisher Full Text
- 88.
Watowich SS:
The erythropoietin receptor: Molecular structure and hematopoietic signaling pathways.
J Investig Med.
2011; 59(7): 1067–72. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 89.
Kumkhaek C, Aerbajinai W, Liu W, et al.:
MASL1 induces erythroid differentiation in human erythropoietin-dependent CD34+ cells through the Raf/MEK/ERK pathway.
Blood.
2013; 121(16): 3216–27. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 90.
Marinkovic D, Zhang X, Yalcin S, et al.:
Foxo3 is required for the regulation of oxidative stress in erythropoiesis.
J Clin Invest.
2007; 117(8): 2133–44. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 91.
Kashii Y, Uchida M, Kirito K, et al.:
A member of Forkhead family transcription factor, FKHRL1, is one of the downstream molecules of phosphatidylinositol 3-kinase-Akt activation pathway in erythropoietin signal transduction.
Blood.
2000; 96(3): 941–9. PubMed Abstract
| Publisher Full Text
- 92.
Liang R, Campreciós G, Kou Y, et al.:
A Systems Approach Identifies Essential FOXO3 Functions at Key Steps of Terminal Erythropoiesis.
PLoS Genet.
2015; 11(10): e1005526. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 93.
Karur VG, Lowell CA, Besmer P, et al.:
Lyn kinase promotes erythroblast expansion and late-stage development.
Blood.
2006; 108(5): 1524–32. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 94.
Schnöder TM, Arreba-Tutusaus P, Griehl I, et al.:
Epo-induced erythroid maturation is dependent on Plcγ 1 signaling.
Cell Death Differ.
2015; 22(6): 974–85. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 95.
Liongue C, Sertori R, Ward AC:
Evolution of Cytokine Receptor Signaling.
J Immunol.
2016; 197(1): 11–8. PubMed Abstract
| Publisher Full Text
- 96.
Li K, Miller C, Hegde S, et al.:
Roles for an Epo Receptor Tyr-343 Stat5 Pathway in Proliferative Co-signaling with Kit.
J Biol Chem.
2003; 278(42): 40702–9. PubMed Abstract
| Publisher Full Text
- 97.
Wu H, Klingmüller U, Besmer P, et al.:
Interaction of the erythropoietin and stem-cell-factor receptors.
Nature.
1995; 377(6546): 242–6. PubMed Abstract
| Publisher Full Text
- 98.
Wessely O, Bauer A, Quang CT, et al.:
A Novel Way to Induce Erythroid Progenitor Self Renewal: Cooperation of c-Kit with the Erythropoietin Receptor.
Biol Chem.
1999; 380(2): 187–202. PubMed Abstract
| Publisher Full Text
- 99.
Wu H, Klingmüller U, Acurio A, et al.:
Functional interaction of erythropoietin and stem cell factor receptors is essential for erythroid colony formation.
Proc Natl Acad Sci U S A.
1997; 94(5): 1806–10. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 100.
Roetto A, Mezzanotte M, Pellegrino RM:
The Functional Versatility of Transferrin Receptor 2 and Its Therapeutic Value.
Pharmaceuticals (Basel).
2018; 11(4): 115. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 101.
Forejtnikovà H, Vieillevoye M, Zermati Y, et al.:
Transferrin receptor 2 is a component of the erythropoietin receptor complex and is required for efficient erythropoiesis.
Blood.
2010; 116(24): 5357–67. PubMed Abstract
| Publisher Full Text
- 102.
Nai A, Lidonnici MR, Rausa M, et al.:
The second transferrin receptor regulates red blood cell production in mice.
Blood.
2015; 125(7): 1170–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 103.
Wortham AM, Goldman DC, Chen J, et al.:
Extrahepatic deficiency of transferrin receptor 2 is associated with increased erythropoiesis independent of iron overload.
J Biol Chem.
2020; 295(12): 3906–17. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 104.
Johnson MB, Enns CA:
Diferric transferrin regulates transferrin receptor 2 protein stability.
Blood.
2004; 104(13): 4287–93. PubMed Abstract
| Publisher Full Text
- 105.
Porpiglia E, Hidalgo D, Koulnis M, et al.:
Stat5 signaling specifies basal versus stress erythropoietic responses through distinct binary and graded dynamic modalities.
PLoS Biol.
2012; 10(8): e1001383. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 106.
Zang H, Sato K, Nakajima H, et al.:
The distal region and receptor tyrosines of the Epo receptor are non-essential for in vivo erythropoiesis.
EMBO J.
2001; 20(12): 3156–66. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 107.
Juvonen E, Ikkala E, Fyhrquist F, et al.:
Autosomal dominant erythrocytosis caused by increased sensitivity to erythropoietin.
Blood.
1991; 78(11): 3066–9. PubMed Abstract
- 108.
Jiao H, Berrada K, Yang W, et al.:
Direct association with and dephosphorylation of Jak2 kinase by the SH2-domain-containing protein tyrosine phosphatase SHP-1.
Mol Cell Biol.
1996; 16(12): 6985–92. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 109.
Baker SJ, Rane SG, Reddy EP:
Hematopoietic cytokine receptor signaling.
Oncogene.
2007; 26(47): 6724–37. PubMed Abstract
| Publisher Full Text
- 110.
Wormald S, Hilton DJ:
Inhibitors of cytokine signal transduction.
J Biol Chem.
2004; 279(2): 821–4. PubMed Abstract
| Publisher Full Text
- 111.
Yoshimura A, Ito M, Chikuma S, et al.:
Negative Regulation of Cytokine Signaling in Immunity.
Cold Spring Harb Perspect Biol.
2018; 10(7): a028571. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 112.
Suessmuth Y, Elliott J, Percy MJ, et al.:
A new polycythaemia vera-associated SOCS3 SH2 mutant (SOCS3F136L) cannot regulate erythropoietin responses.
Br J Haematol.
2009; 147(4): 450–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 113.
Hookham MB, Elliott J, Suessmuth Y, et al.:
The myeloproliferative disorder-associated JAK2 V617F mutant escapes negative regulation by suppressor of cytokine signaling 3.
Blood.
2007; 109(11): 4924–9. PubMed Abstract
| Publisher Full Text
- 114.
Tong W, Zhang J, Lodish HF:
Lnk inhibits erythropoiesis and Epo-dependent JAK2 activation and downstream signaling pathways.
Blood.
2005; 105(12): 4604–12. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 115.
McMullin MF, Cario H:
LNK mutations and myeloproliferative disorders.
Am J Hematol.
2016; 91(2): 248–51. PubMed Abstract
| Publisher Full Text
- 116.
van der Harst P, Zhang W, Mateo Leach I, et al.:
Seventy-five genetic loci influencing the human red blood cell.
Nature.
2012; 492(7429): 369–75. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 117.
McMullin MF, Wu C, Percy MJ, et al.:
A nonsynonymous LNK polymorphism associated with idiopathic erythrocytosis.
Am J Hematol.
2011; 86(11): 962–4. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 118.
Giani FC, Fiorini C, Wakabayashi A, et al.:
Targeted Application of Human Genetic Variation Can Improve Red Blood Cell Production from Stem Cells.
Cell Stem Cell.
2016; 18(1): 73–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 119.
Neumann D, Wikström L, Watowich SS, et al.:
Intermediates in degradation of the erythropoietin receptor accumulate and are degraded in lysosomes.
J Biol Chem.
1993; 268(18): 13639–49. PubMed Abstract
- 120.
Bulut GB, Sulahian R, Ma Y, et al.:
Ubiquitination Regulates the Internalization, Endolysosomal Sorting, and Signaling of the Erythropoietin Receptor.
J Biol Chem.
2011; 286(8): 6449–57. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 121.
Jimenez C, Hernandez C, Pimentel B, et al.:
The p85 regulatory subunit controls sequential activation of phosphoinositide 3-kinase by Tyr kinases and Ras.
J Biol Chem.
2002; 277(44): 41556–62. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 122.
Sulahian R, Cleaver O, Huang LJS:
Ligand-induced EpoR internalization is mediated by JAK2 and p85 and is impaired by mutations responsible for primary familial and congenital polycythemia.
Blood.
2009; 113(21): 5287–97. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 123.
Bulut GB, Sulahian R, Yao H, et al.:
Cbl ubiquitination of p85 is essential for Epo-induced EpoR endocytosis.
Blood.
2013; 122(24): 3964–72. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 124.
Heir P, Srikumar T, Bikopoulos G, et al.:
Oxygen-dependent Regulation of Erythropoietin Receptor Turnover and Signaling.
J Biol Chem.
2016; 291(14): 7357–72. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 125.
Khalil S, Delehanty L, Grado S, et al.:
Iron modulation of erythropoiesis is associated with Scribble-mediated control of the erythropoietin receptor.
J Exp Med.
2018; 215(2): 661–79. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 126.
Broxmeyer HE, Hoggatt J, O'Leary HA, et al.:
Dipeptidylpeptidase 4 negatively regulates colony-stimulating factor activity and stress hematopoiesis.
Nat Med.
2012; 18(12): 1786–96. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 127.
Vainchenker W, Leroy E, Gilles L, et al.:
JAK inhibitors for the treatment of myeloproliferative neoplasms and other disorders [version 1; peer review: 2 approved].
F1000Res.
2018; 7: 82. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 128.
Huang LJ, Constantinescu SN, Lodish HF:
The N-Terminal Domain of Janus Kinase 2 Is Required for Golgi Processing and Cell Surface Expression of Erythropoietin Receptor.
Mol Cell.
2001; 8(6): 1327–38. PubMed Abstract
| Publisher Full Text
- 129.
Yao H, Ma Y, Hong Z, et al.:
Activating JAK2 mutants reveal cytokine receptor coupling differences that impact outcomes in myeloproliferative neoplasm.
Leukemia.
2017; 31(10): 2122–31. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 130.
Wilmes S, Hafer M, Vuorio J, et al.:
Mechanism of homodimeric cytokine receptor activation and dysregulation by oncogenic mutations.
Science.
2020; 367(6478): 643–52. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 131.
Constantinescu SN, Huang LJ, Nam H, et al.:
The Erythropoietin Receptor Cytosolic Juxtamembrane Domain Contains an Essential, Precisely Oriented, Hydrophobic Motif.
Mol Cell.
2001; 7(2): 377–85. PubMed Abstract
| Publisher Full Text
- 132.
Ferrao RD, Wallweber HJ, Lupardus PJ:
Receptor-mediated dimerization of JAK2 FERM domains is required for JAK2 activation.
eLife.
2018; 7: e38089. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 133.
Kim AR, Ulirsch JC, Wilmes S, et al.:
Functional Selectivity in Cytokine Signaling Revealed Through a Pathogenic EPO Mutation.
Cell.
2017; 168(6): 1053–1064.e15. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 134.
Mohan K, Ueda G, Kim AR, et al.:
Topological control of cytokine receptor signaling induces differential effects in hematopoiesis.
Science.
2019; 364(6442): eaav7532. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 135.
Wootten D, Christopoulos A, Marti-Solano M, et al.:
Mechanisms of signalling and biased agonism in G protein-coupled receptors.
Nat Rev Mol Cell Biol.
2018; 19(10): 638–53. PubMed Abstract
| Publisher Full Text
- 136.
Thomas C, Moraga I, Levin D, et al.:
Structural linkage between ligand discrimination and receptor activation by type I interferons.
Cell.
2011; 146(4): 621–32. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 137.
McKeithan TW:
Kinetic proofreading in T-cell receptor signal transduction.
Proc Natl Acad Sci U S A.
1995; 92(11): 5042–6. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 138.
Ng T, Marx G, Littlewood T, et al.:
Recombinant erythropoietin in clinical practice.
Postgrad Med J.
2003; 79(933): 367–76. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 139.
Mikhail A, Brown C, Williams JA, et al.:
Renal association clinical practice guideline on Anaemia of Chronic Kidney Disease.
BMC Nephrol.
2017; 18(1): 345. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 140.
van Swelm RPL, Wetzels JFM, Swinkels DW:
The multifaceted role of iron in renal health and disease.
Nat Rev Nephrol.
2020; 16(2): 77–98. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 141.
Ganz T, Nemeth E:
Iron Balance and the Role of Hepcidin in Chronic Kidney Disease.
Semin Nephrol.
2016; 36(2): 87–93. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 142.
Won HH, Park I, Lee E, et al.:
Comparative analysis of the JAK/STAT signaling through erythropoietin receptor and thrombopoietin receptor using a systems approach.
BMC Bioinformatics.
2009; 10(Suppl 1): S53. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 143.
Shi J, Yuan B, Hu W, et al.:
JAK2 V617F stimulates proliferation of erythropoietin-dependent erythroid progenitors and delays their differentiation by activating Stat1 and other nonerythroid signaling pathways.
Exp Hematol.
2016; 44(11): 1044–1058.e5. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 144.
Funakoshi-Tago M, Sumi K, Kasahara T, et al.:
Critical Roles of Myc-ODC Axis in the Cellular Transformation Induced by Myeloproliferative Neoplasm-Associated JAK2 V617F Mutant.
PLoS One.
2013; 8(1): e52844. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 145.
Cai X, Jia H, Liu Z, et al.:
Polyhydroxylated fullerene derivative C60(OH)24 prevents mitochondrial dysfunction and oxidative damage in an MPP+ -induced cellular model of Parkinson's disease.
J Neurosci Res.
2008; 86(16): 3622–34. PubMed Abstract
| Publisher Full Text
- 146.
Funakoshi-Tago M, Nagata T, Tago K, et al.:
Fullerene derivative prevents cellular transformation induced by JAK2 V617F mutant through inhibiting c-Jun N-terminal kinase pathway.
Cell Signal.
2012; 24(11): 2024–34. PubMed Abstract
| Publisher Full Text
- 147.
Eschbach JW, Aquiling T, Haley NR, et al.:
The long-term effects of recombinant human erythropoietin on the cardiovascular system.
Clin Nephrol.
1992; 38 Suppl 1: S98–103. PubMed Abstract
- 148.
Guglin ME, Koul D:
Cardiovascular effects of erythropoietin: Anemia and beyond.
Cardiol Rev.
2006; 14(4): 200–4. PubMed Abstract
| Publisher Full Text
- 149.
Gupta N, Wish JB:
Erythropoietin and its cardiovascular effects. In: Rangaswami J, Lerma EV, Ronco C, editors. Cardio-Nephrology. Cham: Springer; 2017; 119–28. Publisher Full Text
- 150.
Casati S, Passerini P, Campise MR, et al.:
Benefits and risks of protracted treatment with human recombinant erythropoietin in patients having haemodialysis.
Br Med J (Clin Res Ed).
1987; 295(6605): 1017–20. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 151.
Debeljak N, Solár P, Sytkowski AJ:
Erythropoietin and cancer: The unintended consequences of anemia correction.
Front Immunol.
2014; 5: 563. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 152.
Jelkmann W:
Physiology and Pharmacology of Erythropoietin.
Transfus Med Hemother.
2013; 40(5): 302–9. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 153.
Pradeep S, Huang J, Mora EM, et al.:
Erythropoietin Stimulates Tumor Growth via EphB4.
Cancer Cell.
2015; 28(5): 610–22. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 154.
Remuzzi G, Ingelfinger JR:
Correction of anemia--payoffs and problems.
N Engl J Med.
2006; 355(20): 2144–6. PubMed Abstract
| Publisher Full Text
- 155.
Singh AK, Szczech L, Tang KL, et al.:
Correction of anemia with epoetin alfa in chronic kidney disease.
N Engl J Med.
2006; 355(20): 2085–98. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 156.
Pfeffer MA, Burdmann EA, Chen CY, et al.:
A Trial of Darbepoetin Alfa in Type 2 Diabetes and Chronic Kidney Disease.
N Engl J Med.
2009; 361(21): 2019–32. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 157.
Miller CP, Lowe KA, Valliant-Saunders K, et al.:
Evaluating erythropoietin-associated tumor progression using archival tissues from a phase III clinical trial.
Stem Cells.
2009; 27(9): 2353–61. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 158.
Elliott S, Sinclair AM:
The effect of erythropoietin on normal and neoplastic cells.
Biologics.
2012; 6: 163–89. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 159.
Macdougall IC, White C, Anker SD, et al.:
Intravenous Iron in Patients Undergoing Maintenance Hemodialysis.
N Engl J Med.
2019; 380(5): 447–58. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 160.
Bohlius J, Bohlke K, Castelli R, et al.:
Management of cancer-associated anemia with erythropoiesis-stimulating agents: ASCO/ASH clinical practice guideline update.
Blood Adv.
2019; 3(8): 1197–210. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 161.
Platzbecker U, Symeonidis A, Oliva EN, et al.:
A phase 3 randomized placebo-controlled trial of darbepoetin alfa in patients with anemia and lower-risk myelodysplastic syndromes.
Leukemia.
2017; 31(9): 1944–50. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 162.
Park S, Greenberg P, Yucel A, et al.:
Clinical effectiveness and safety of erythropoietin-stimulating agents for the treatment of low- and intermediate-1-risk myelodysplastic syndrome: A systematic literature review.
Br J Haematol.
2019; 184(2): 134–60. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 163.
Steensma DP:
Hematopoietic Growth Factors in Myelodysplastic Syndromes.
Semin Oncol.
2011; 38(5): 635–47. PubMed Abstract
| Publisher Full Text
- 164.
Whoriskey P:
The rise and fall of a billion-dollar drug. Washington Post. 2012. Reference Source
- 165.
Kaplan J:
Roxadustat and Anemia of Chronic Kidney Disease.
N Engl J Med.
2019; 381(11): 1070–2. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 166.
Sanghani NS, Haase VH:
Hypoxia-Inducible Factor Activators in Renal Anemia: Current Clinical Experience.
Adv Chronic Kidney Dis.
2019; 26(4): 253–66. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 167.
Chen N, Hao C, Liu BC, et al.:
Roxadustat Treatment for Anemia in Patients Undergoing Long-Term Dialysis.
N Engl J Med.
2019; 381(11): 1011–22. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 168.
Brigandi RA, Johnson B, Oei C, et al.:
A Novel Hypoxia-Inducible Factor−Prolyl Hydroxylase Inhibitor (GSK1278863) for Anemia in CKD: A 28-Day, Phase 2A Randomized Trial.
Am J Kidney Dis.
2016; 67(6): 861–71. PubMed Abstract
| Publisher Full Text
- 169.
Besarab A, Provenzano R, Hertel J, et al.:
Randomized placebo-controlled dose-ranging and pharmacodynamics study of roxadustat (FG-4592) to treat anemia in nondialysis-dependent chronic kidney disease (NDD-CKD) patients.
Nephrol Dial Transplant.
2015; 30(10): 1665–73. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 170.
Provenzano R, Besarab A, Sun CH, et al.:
Oral Hypoxia–Inducible Factor Prolyl Hydroxylase Inhibitor Roxadustat (FG-4592) for the Treatment of Anemia in Patients with CKD.
Clin J Am Soc Nephrol.
2016; 11(6): 982–91. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 171.
Dhillon S:
Roxadustat: First Global Approval.
Drugs.
2019; 79(5): 563–72. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 172.
Ma S, Chen J, Chen C, et al.:
Erythropoietin Rescues Memory Impairment in a Rat Model of Chronic Cerebral Hypoperfusion via the EPO-R/JAK2/STAT5/PI3K/Akt/GSK-3β Pathway.
Mol Neurobiol.
2018; 55(4): 3290–9. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 173.
Kimáková P, Solár P, Solárová Z, et al.:
Erythropoietin and Its Angiogenic Activity.
Int J Mol Sci.
2017; 18(7): 1519. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 174.
Broxmeyer HE:
Erythropoietin: Multiple targets, actions, and modifying influences for biological and clinical consideration.
J Exp Med.
2013; 210(2): 205–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 175.
Brines M, Grasso G, Fiordaliso F, et al.:
Erythropoietin mediates tissue protection through an erythropoietin and common beta-subunit heteroreceptor.
Proc Natl Acad Sci U S A.
2004; 101(41): 14907–12. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 176.
Razak A, Hussain A:
Erythropoietin in perinatal hypoxic-ischemic encephalopathy: A systematic review and meta-analysis.
J Perinat Med.
2019; 47(4): 478–89. PubMed Abstract
| Publisher Full Text
| Faculty Opinions Recommendation
- 177.
Tsai TH, Lu CH, Wallace CG, et al.:
Erythropoietin improves long-term neurological outcome in acute ischemic stroke patients: A randomized, prospective, placebo-controlled clinical trial.
Crit Care.
2015; 19(1): 49. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
- 178.
Nekoui A, Blaise G:
Erythropoietin and Nonhematopoietic Effects.
Am J Med Sci.
2017; 353(1): 76–81. PubMed Abstract
| Publisher Full Text
- 179.
Suresh S, Rajvanshi PK, Noguchi CT:
The Many Facets of Erythropoietin Physiologic and Metabolic Response.
Front Physiol.
2020; 10: 1534. PubMed Abstract
| Publisher Full Text
| Free Full Text
| Faculty Opinions Recommendation
Comments on this article Comments (0)