Keywords
SARS-CoV-2, Molecular docking, COVID-19
This article is included in the Emerging Diseases and Outbreaks gateway.
This article is included in the Cheminformatics gateway.
This article is included in the Coronavirus (COVID-19) collection.
SARS-CoV-2, Molecular docking, COVID-19
Since 2003, three coronaviruses have been associated with pneumonia, the first was severe acute respiratory syndrome coronavirus (SARS-CoV-1)1 which affected 8,098 people causing 774 deaths between 2002 and 20032, the second was Middle-East respiratory syndrome coronavirus (MERS-CoV)3 and the third is SARS-CoV-2 which, as at 8th July, 2020, has affected 11,669,259 globally and is responsible for 539,906 deaths. SARS-CoV-2 is a human pathogen which has been declared a global pandemic by the World Health Organization4 and is responsible for the Coronavirus disease 2019 (COVID-19) (1). . The bats have been identified as the possible reservoir and origin for both the SARS-CoV-2 and SARS-COV-13. Unfortunately, there has been no known cure for COVID-19 to date.3
The entry into the host cell by the coronaviruses is usually mediated by the spike (S) glycoprotein3. This glycoprotein interacts with the angiotensin-converting enzyme 2 (ACE2), enabling the virus penetration into the host. The main protease (M protease, also known as 3CL protease) has also been found to be essential for processing of translated polyproteins for the SARS_CoV-2 virus. The two targets, the S glycoprotein, Sgp and MPro proteins have therefore been considered as important drug targets against the SARS-CoV-2 virus.5. For this study, these two drug targets were selected and used to virtually screen some phytochemicals for possible activity against the SARS-CoV-2 virus.
Possibly, potent inhibitors of these two targets will be able to interfere with the SARS-CoV-2 replication process and thus serves as potential drugs for the management of the COVID-19. Hence, this work is aimed at identifying some potential lead compounds of plant origin that can serve as candidates for testing against the SARS-CoV-2 virus.
Plant compounds reported in the literatures that have been demonstrated to have antiviral activities (list of all the compounds analyzed is available as Extended data6) were selected, alongside also hydroxychloroquine, remdesivir and favipiravir, which have been used in the treatment and management of COVID-19 and mined from the PubChem database.
Two proteins including crystal structure of the SARS-CoV-2 main protease (PDB ID 6LU7)7 in complex with an inhibitor N3 and the S glycoprotein in complex with N-acetyl-D-glucosamine (NAG) (PDB ID 6VSB) were downloaded from the Protein Data Bank (www.rcsb.org). The proteins were prepared by removing water molecules and co-crystalized ligands (highlighting the water molecules and co-crystalized ligands and deleting) on the proteins using Discovery Studio version 2017R2 (19). The UCSF Chimera molecular modeling package is an open access equivalent that could be sued to perform the same function8.
The already prepared protein and ligands were loaded on to the PyRx docking software (PyRx-Python Prescription 0.8), where molecular docking was done in the AutodockVina mode. Visualization to see how the ligands fitted and bound into the binding pockets on the protein and also the interactions between the protein and the ligands was done using Discovery Studio version 2017R2 (19) by first loading the saved PDBqt output file of the target protein from PyRx and then inserting the output of the binding modes of the different ligands and then viewed under the Receptor-Ligand interaction platform of Discovery Studio software.
The Physicochemical properties and drugability of selected compounds were predicted using the free online versions of SwissADME9 and Molinspiration10 platforms and their predicted toxicity profile also compared using the PROTOX toxicity prediction platform11. In each case the ligands were loaded onto the platforms as SMILES structures obtained from PubChem.
A library of 22 compounds of plant origin known to have antiviral activity was obtained from the PubChem database (see Extended data6). Though the compounds are chemically diverse, they consist of largely flavonoids and terpenes. Some compounds from the citrus family were found among the library, though they could not make it among the top six selected compounds that demonstrated good binding affinities for the two targets. Most of the compounds has showed similar binding affinities to the selected protein targets (6LU7 (M protease) and 6VSB (S glycoprotein)) compared to the training sets of known ligands to the selected targets (see Table 1).
However, the top six compounds with most favorable binding affinity were selected for each of the targets. The outcomes of the binding affinities of the selected compounds on the 6LU7 and 6VBS targets are presented in Table 2 and Table 3, respectively.
The binding affinities of the top six compounds (Table 1) on the 6vsb target are comparable to each other, i.e. they all lie within a close range of 9 to 9.6 kcal/mol indicating that they might likely have equal or comparable potential as lead compounds for the 6vsb S glycoprotein.
One of the compounds sylibinin12 is an FDA approved drug, which showed up as active on both M protease and S glycoprotein will make a good candidate of repurposing. Finding Quercetin as a potential inhibitor of the M protease Protein (6LU7) of SARS-CoV-2 corresponds with an earlier report13.
Looking at the octanol–water coefficient (cLogP) of the compounds, there was no correlation observed between the lipophilicity and the interaction with the receptors. However, for the compounds acting on 6LU7 (Serial numbers 3, 4, 7, 8, 9 and 10 in Table 4), interaction with the receptor is correlated with low lipophilicity, with the exception of solanidine and dammarenolic acid, which have high cLogP values, although both compounds also use their polar functional groups in interacting with the receptor. Bacailin (Figure 1) and naringenin showed good hydrogen bond interaction with the 6VSB receptor due to their polarity.
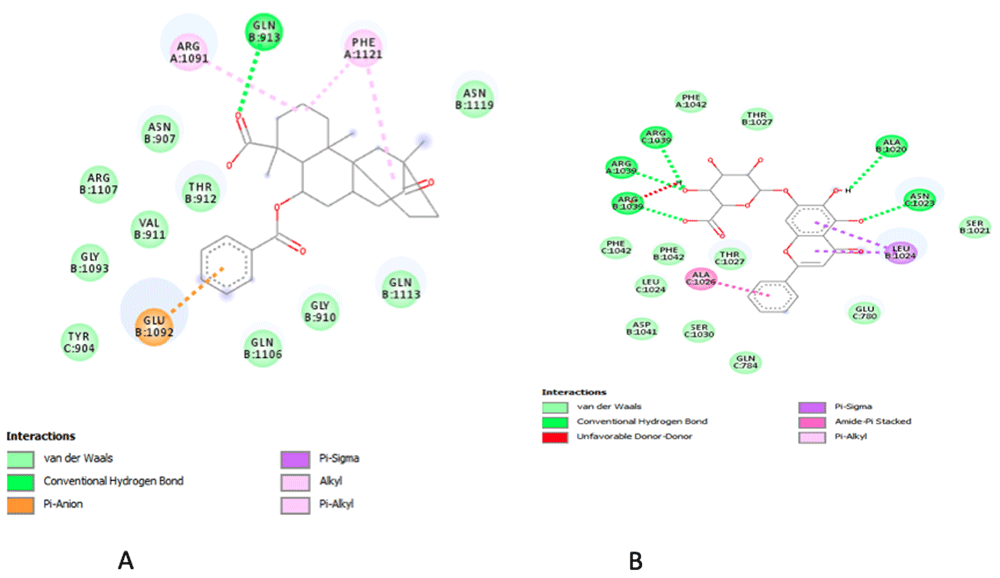
Best binding pose and interaction of (A) scopodulcic acid and (B) baicalin on the spike glycoprotein.
Filtering the compounds for drug likeness on the basis of Linpinski’s and/or Veber’s rule showed that all the compounds have drug-like properties except baicalin, which failed the two filtering scales applied (Table 5). This implies that baicalin is not worth considering further without any structural modification. The predicted toxicity profile of the selected compounds shows (Table 6) that all the compounds are likely to be relatively safe, which makes them good potential candidates for anti-infectives because the chances of achieving selective toxicity is high. Baicalin is thus most likely the safest.
Two compounds among the top six selected for each target, solanidine and sylibinin, were observed to have good binding affinity on both the 6VSB and the 6FLU7 proteins. This makes them potential multitarget acting inhibitors on the SARS-CoV-2. Solanidine is a steroidal glycoalkaloid found in potatoes14. Although toxic to humans and animals, solanidine has been reported to be effective against herpes viruses (HSV), herpes genitalis and herpes zoster15 Its activity against HSV is attributed to the presence of a sugar moiety16. In silico drug screening using PROTOX II showed that solanidine is very likely to be cytotoxic and immunotoxic. PROTOTOX II is a cost- and time-saving approach for testing and determining the toxicity of a compound to be considered a drug of choice17. PROTOTOX II predicts the toxicity outcome of a potential drug of choice, it incorporates machine-learning models which use a combination of fragment propensities, molecular similarity, pharmacophores, to predict toxicity endpoints, such as acute toxicity, cytotoxicity, , carcinogenicity , hepatotoxicity, immunotoxicity, mutagenicity and toxicity targets17
A safe drug must not be toxic to its host target. Based on the PROTOX II evaluation of toxicity, dammarenolic acid emerges as the compound of choice with the least toxicity. Dammarenolic acid has been reported as effective antiviral agents dammarenolic acid potently inhibited the in vitro replication of other retroviruses, including simian immunodeficiency virus and murine leukemic virus in vector-based antiviral screening studies and has been proposed as a potential lead compound in the development of anti-retrovirals18. The compound is cytotoxic and demonstrate potential against respiratory syncytial virus19. We therefore propose that the evaluation of dammarenolic acid might hold the key to a safe and effective anti-SARS-CoV-2 drug considering its drugability and low toxicity.
This study proposes a potential re-purposing of silybinin for the management of COVID19 diseases. Silybinin (silymarin) possesses antiviral ability against hepatitis C virus (HCV)20,21 It has been reported to have activities against a wide range of viral groups including flaviviruses (HCV and dengue virus), togaviruses (Chikungunya virus and Mayaro virus), influenza virus, human immunodeficiency virus, and hepatitis B virus20. In an in vivo and in vitro study, Silymarin has been proposed to inhibit HCV entry, RNA synthesis, viral protein expression and prevent infectious virus production; it can also block cell-to-cell spread of the virus22. In silico analysis of silybinin in this present study has shown that it can likely inhibit SARS-CoV-2 S glycoprotein and Mpro targets, making it a drug to be considered with a possible multi-target activity against the SARS-CoV-2 virus.
From the 22 phytocompounds that were virtually screened, scopodulcic acid and dammarenolic acid showed the best binding energies with the S glycoprotein and Mpro, respectively. This makes them potential lead compounds for development into candidates against the SARS-CoV-2. Furthermore, the FDA-approved drug silybinin (Legalon) had good binding affinity for the two targets, so could be evaluated further for possible repurposing against the SARS-CoV-2 virus.
All data underlying the results are available as part of the article and no additional source data are required.
Harvard Dataverse: Replication Data for:Molecular docking analysis of some phytochemicals on two SARS-CoV-2 targets. https://doi.org/10.7910/DVN/BB0QQK6.
The file within this project contains the compounds obtained from the PubChem database that were analyzed in this study.
Extended data are available under the terms of the Creative Commons Zero "No rights reserved" data waiver (CC0 1.0 Public domain dedication).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)