Keywords
somatotransgenic, luciferase, germline transgenic, biosensor, lentivirus, AAV
This article is included in the NC3Rs gateway.
somatotransgenic, luciferase, germline transgenic, biosensor, lentivirus, AAV
Reduced number of animals (a cohort of approximately 16 animals for multiple time points across the entire experiment compared to 16 animals PER time point); more data from same number of animals as serial measures can be performed; refined techniques to allow for conscious imaging; reduced anesthesia events which decreases animal distress leading to improved welfare of the animals.
In the past, the study of gene expression during normal development or induced by disease in rodent models have been assessed largely by end-point assays involving the sacrifice of large cohorts of animals or restricted to invasive serial blood or urine sampling. Whole body imaging has been a game changer in this context and bioluminescence imaging (BLI) has become one of the most prevalent non-invasive modalities in academic research. Luciferase enzymes, derived from many different invertebrate species, catalyze their substrates in a reaction that emits photons of light in a dose responsive and quantifiable manner1. Bioluminescent light, resulting from the activity of luciferase genes optimized for expression in mammalian cells, can be used as a quantifiable surrogate for gene promoter activation in vivo and detected using highly sensitive charge-coupled device (CCD) cameras2. Germline, light-producing transgenic (LPT) mice, where a luciferase transgene is inserted in the first intron of an endogenous gene, can be subjected to BLI to quantify gene activity in living animals3. Genome editing technologies have expedited the generation of whole body luciferase reporter transgenics4, and have massively increased the usage of LPT rodents, but critically do not reduce animal wastage in transgenic colony formation. In fact, the numbers of mice used in biomedical research has increased substantially since CRISPR/Cas9 genome editing has been applied to the production of transgenic mice with the time taken for mouse models to be created taking months instead of years to generate. The UK Home Office statistics notes that the number of procedures involving genetically altered animals rose by 36% between 2007 and 2016. Furthermore, it was reported that in 2016, 226,000 animals were used to create new lines of genetically altered animals5.
One of the greatest challenges facing users of germline LPT mice is that the luciferase reporter is present in every cell of the mouse. Researchers studying gene activity in a particular tissue or organ must be able to differentiate between target bioluminescence and the surrounding noise or targeting of the signal from a different organ entirely6. For many visceral organs this is practically impossible. We have developed an alternative approach by targeting conditionally activated luciferase reporter cassettes using viral vectors (lentivirus and adeno-associated virus) administered directly to target tissues in the newborn rodent to produce tissue-restricted somatotransgenic animals2. Viral transduction is further targeted by the use of pseudotyping with alternative viral envelopes known to have a specific cellular or tissue tropism7. Resultant somatotransgenic rodents have tissue-restricted expression of a luciferase gene conditionally activated by a minimally defined promoter or a synthetic promoter. To date, we have designed and validated in vitro over 20 synthetic transcription factor activated reporter (TFAR) constructs based on a minimal promoter sequence activated by serial transcription factor binding motifs2. These reporter elements have been selected to interrogate some of the most common signaling pathways known to be aberrant in neurological diseases, cancer, fibrosis and drug toxicity, the details of which are highlighted in Table 1. Any small rodent can be subjected to somatotransgenic bioimaging (SB) and there is no colony breeding wastage.
nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB); lipopolysaccharide (LPS); bone morphogenetic protein 2a (BMP2a); T-cell factor/lymphoid enhancer-binding factor (TCF/LEF); lithium chloride (LiCl2); activator protein 1 (AP-1); phorbol myristate acetate (PMA); Leukemia inhibitory factor (LIF); hepatic nuclear factor 4 beta (HNF4-β); hydrogen peroxide (H2O2); 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD); nuclear factor of activated T-cells (NFAT); Notch intracellular domain (NICD); liver x receptor (LXR); transcription factor EB (TFEB).
Furthermore, SB can be layered on either gene knockout, chemically/biologically induced or naturally occurring rodent disease models2. The combined flexibility and reliability of SB means that the numbers of animals required for experiments is substantially reduced. As an example, a literature review of over 20 comparable studies of liver disease revealed that the common analytical methodologies were serial blood, bile and urine sampling involving repeated anaesthesia as well as periodic sacrifice for histological analysis. These involved large cohorts of between 3-12 animals being sacrificed at up to 10 timepoints meaning as many as 120 animals are used in each experiment. Typically, our experiments with somatotransgenic animals and longitudinal imaging contains 6-8 experimental and 6-8 control animals per experiment. This translates into a 7-10 fold reduction in the number of animals used as compared to the current standard methodologies of analysis.
It is established that repeated anesthesia is not only distressing to rodents but also disrupts physiological and pathological processes, notably in the brain8. BLI is routinely carried out on anesthetized mice in order to define a photonic capture region of interest on a static target. Since SB is based on complete capture from a tissue-restricted luciferase reporter gene, rodents can remain conscious and unrestrained during imaging. Our comparisons of SB in brains and livers showed no difference between conscious or unconscious bioimaging9. This represents a significant refinement to animal husbandry, as well as increases the frequency with which rodents can be subjected to bioimaging. Thus meaning more data can be obtained from the same number of animals. SB can be used by any researcher in academia or industry with access to an appropriate in vivo imaging system, such as the IVIS® imaging platform, to detect BLI and assess changes in gene expression resulting from disease onset or drug intervention.
Here we present methodologies describing the development of the viral reporter vectors, the production of TFAR-containing virus, and methods for in vitro validation. Technical details of our in vivo validations for 2 disease models of inflammation, bile duct ligation and a model of hyperoxia, are detailed and experimental data presented.
MluI (New England Biolabs; R0198S)
XhoI (New England Biolabs; R0146S)
BamHI (New England Biolabs; R0136S)
EcoRI (New England Biolabs; R0101S)
pENTR-1A® (Invitrogen; A10462)
cPPT forward sequencing primer - (GTGCAGGGGAAAGAATAGTAG)(Sigma-Aldrich)
Gateway™ LR Clonase™ II enzyme mix (Invitrogen; 11791020)
DH5α chemically competent cells (Invitrogen; 18258012)
One Shot™ Stbl3™ competent cells (Invitrogen; C737303)
ampicillin antibiotic (Sigma-Aldrich; A9393)
LB agar (Sigma-Aldrich; L3027)
LB broth powder (Sigma-Aldrich; L3147)
agarose gel powder (Sigma-Aldrich; A4718)
HEK293T cell line (VWR; MSPP-CRL3216)
Dulbecco’s Modified Eagle’s Media (DMEM) (Sigma-Aldrich; D6429)
Fetal calf serum (FCS)(Invitrogen; 10082147)
Penicillin/Streptomycin (Pen/Strep) (Sigma-Aldrich; P4333)
Phosphate buffered saline (PBS) (Sigma-Aldrich; D8662)
Trypsin EDTA dissociation media (Sigma-Aldrich; T4299)
Opti-MEM I reduced serum medium (Invitrogen; 31985070)
pMD2.G (VSV-G envelope) (Addgene; #12259)
pCMVd8.74 (gag-pol, tat, rev) (Addgene; #22036)
PsPAX2 (Addgene #12260)
RETRO-TEK HIV-1 p24 antigen ELISA (Zeptometrix; 0801200)
KAPA SYBR FAST qPCR kit (Roche; KK4600)
branched PEI (MW ~25,000) (lentivirus production) (Sigma Aldrich; #408727)
0.45 µM PVDF filter (Sigma-Aldrich; P1938)
pHGT1 Ad5 helper plasmid (MTA Harvard Medical School, USA)
Benzonase (Sigma-Aldrich; E8263)
AAV pro 293T cells (Takara; 632273)
Sodium chloride (Sigma-Aldrich; S9888)
Polyethyleneimine (PEI) (Polysciences; 24765)
0.45 µm filter membrane (Millipore; SCHVU05RE)
Cell lifter (Corning; 3008)
0.45 µm syringe filters (Sartorius; 17598)
0.22 µm centrifuge tube filter (Costar; 8160)
10 ml syringe (Terumo; SS+10ES1)
50 ml syringe (BD Plastipak; 300865)
Disposable needles (BD Microlance; 301155)
Benzonase nuclease (Sigma; E1014-25KU)
Amicon Ultra-15 centrifugal filter units (Millipore; UFC910024)
Slide-A-lyzer dialysis cassette 10,000 MWCO (Thermo Scientific; 66810)
5 ml FACS tubes (Falcon; 352053)
Sodium deoxycholate (Sigma; D6750-100G)
Glycine (Sigma; G8898-500G)
POROS™ CaptureSelect™ AAV Resin (Thermo Scientific; A36739)
ÄKTAprime plus (High performance liquid chromatography - HPLC system (GE Healthcare; 11001313)
DPBS (1X) (Gibco; 14190-094)
PBS tablets (Sigma; P4417-100TAB)
Luna Universal Probe qPCR MasterMix (NEB; M3004S).
96-well PCR plate 0.1 mL format.
MicroAmp® Optical Adhesive Film (Applied Biosystems).
HeLa cell line (Sigma; 93021013)
Huh7 cell line (JCRB Cell Bank; JCRB0403)
HepG2 cell line (Sigma; 85011430)
RPMI-1640 media (Sigma-Aldrich; R8758)
ultra-pure LPS-EB (InvivoGen; tlrl-3pelps)
Activin A (PeproTech; AF-120-14E)
BMP2a (PeproTech; AF-120-02)
Cobalt Chloride (Sigma-Aldrich; 60818)
Lithium Chloride (LiCl2) (Sigma-Aldrich; L9650)
Estradiol (Sigma-Aldrich; E2758)
Nutlin-3a (Sigma-Aldrich; SML0580)
Phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich; P8139)
LY294002 (StemCell Technologies; #72152)
human recombinant LIF (StemCell Technologies; #78055)
Purmorphamine (Sigma-Aldrich; SML0868)
Hydrogen peroxide (H2O2) (Sigma-Aldrich; 216763)
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) (Sigma-Aldrich; NIST1614)
Ionomycin (Sigma-Aldrich; I0634)
Luciferase assay lysis buffer (0.65% NP40 (Sigma-Aldrich; NP40), 10 mM Trizma® base (Sigma-Aldrich; T1699) (pH 8.0), 1 mM Ethylenediaminetetraacetic acid (EDTA) (Sigma-Aldrich; E9884)(pH 8.0), 150 mM NaCl(Sigma-Aldrich; S9888))
luciferase assay buffer (25 mM Trizma® base (Sigma-Aldrich; T1699) pH 7.8, 1 mM 1,4-Dithiothreitol (DTT) (Sigma-Aldrich; 10197777001), 1 mM EDTA (Sigma-Aldrich; E9884), 1% Triton™ X-100 (Sigma-Aldrich;), 8 mM MgCl2(Sigma-Aldrich; M8266), 3 ml glycerol (Sigma-Aldrich; G5516), 1.25 mM rATP (Promega; E6011), 0.5% BSA (Sigma-Aldrich; A2153))
Luminometer such as the GloMax (Promega; E9032)
Bio-rad protein assay (Bio-rad; #5000006)
CD1 mice (Charles River Laboratories)
4% isofluorane (Abbott Laboratories)
33-gauge Hamilton needle (Fisher Scientific, Loughborough)
bupivacaine hydrochloride (Advanz Pharma)
D-luciferin, potassium salt (Gold Biotechnologies; LUCK)
cooled charged-coupled device camera (IVIS machine) (Perkin Elmer)
Living Image Software (v4.1) (Perkin Elmer)
Lentivirus cloning. A simple two-step cloning process was developed to generate lentiviral TFAR vectors using Gateway® cloning. We first generated the parental lentiviral vector by cloning the Gateway® (GW) Destination recombination site into a blunted unique MluI restriction site of a 2nd generation HIV-1 based lentiviral backbone upstream of a bicistronic 3xFLAG-FLuc-F2A-eGFP reporter gene cassette to produce pLNT-GW-Luc-eGFP. We then generated a shuttle vector containing the adenovirus E1A minimal promoter (MP) sequence cloned into a unique XhoI restriction site in the Gateway® shuttle vector, pENTR-1A. This shuttle plasmid (pENTR-MP) was used as a sub-cloning vector to introduce de novo synthesized regulatory sequences (Aldevron, North Dakota, USA) upstream of the MP (existing TFARs listed in Table 1) into BamHI/EcoRI restriction sites. Vectors were confirmed by Sanger sequencing using a cPPT forward primer (GTGCAGGGGAAAGAATAGTAG) then recombined into pLNT-GW-Luc-eGFP using Gateway® cloning (Figure 1A).
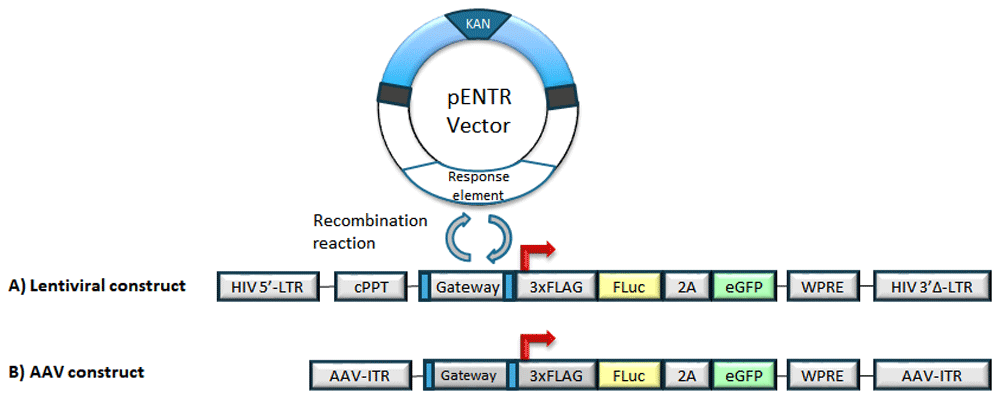
Response elements are cloned into the pENTR vector. A recombination reaction is performed to shuttle the response element into the lentiviral or AAV backbone upstream of a 3xFLAG-FLuc-2A-eGFP bicistronic reporter.
AAV cloning. The AAV8-Gateway®-Luc-T2A-eGFP was completely de novo synthesized (Aldevron). This AAV plasmid consisted of a Gateway® sequence, placed upstream of the Luc-2A-eGFP reporter cassette. AAV TFARs were generated by Gateway® cloning as with the lentiviral vectors (Figure 1B).
Both lentivirus and AAV vectors were generated by transfection of human embryonic kidney, HEK293T, producer cells with accessory plasmids as previously detailed2. Lentiviral titer was generated based on the p24 ELISA assay whereas AAV was titered by quantitation of capsid protein by viral genome copy numbers per ml10,11.
All newly generated TFARs were comprehensively evaluated for predicted responses to known activators/inhibitors on multiple cell lines, including HEK293T, HeLa, Huh7, HepG2, and primary human fibroblasts, prior to being employed in vivo2. The reporters, in vitro agonists, concentrations, and duration of activation are outlined in Table 1.
Specifically, for the HIF response element (HRE) construct that can be used to determine the induction of hypoxia, HEK293T and HeLa cells were transduced with LNT-HRE-Luc-eGFP at a multiplicity of infection (MOI) of 10. Three days post-transduction, transduced cells were replated in two sets of triplicates, one for HRE activation and one for basal levels of luciferase activity. The following day one triplicate set of cells were exposed to the agonist, CoCl2 (100 mM/µl), in RPMI-1640 for 12 hours while the non-activated cells were only placed in RPMI-1640. A time course of 12, 24, 48, and 72 hours post agonist activation was performed. At each time point, the cells were lysed in a standard luciferase assay lysis buffer and stored for future luciferase assay analysis.
All procedures were performed under United Kingdom Home Office Project License 70/8030, approved by the ethical review committee, and followed institutional guidelines at University College London. All methods were performed in accordance with the relevant guidelines and regulations. Male and female outbred wild type CD1 mice were used and were supplied by Charles Rivers Laboratories. Mice were used for adult interventions as close as possible to 6 weeks of age and 25g.
IVC cages were used to house the mice, with a maximum of 5 per cage. Male and female mice were housed separately post weaning. Food and water were routinely monitored by the animal technicians at the facility. The temperature of the room was set at 25°C with light/dark cycles alternating every 12 hrs. Mice were bred onsite and time-mated in order to plan experiments as accurately as possible and avoid wastage of unused pups. A total of 34 mice were used in the studies, described below.
Neonatal intracranial and intravenous injections. Mice either received intracranial or intravenous injections, never both. Skilled technicians can achieve more than 99% success rate for intravenous injections and no failed intracranial injections. No anaesthesia was used as the procedure is very quick and involves a single needle puncture per mouse. For intracranial injections, mice (postnatal day 1 (P1)) received unilateral injections of concentrated lentiviral or AAV vector (5 μl in total) into the cerebral lateral ventricles using a 33-gauge Hamilton needle. The site of injection was located two-fifths of the distance between the eye and the lambda intersection of the skull as described and illustrated by Kim et al.12. During the procedure, mice were held in an upright position, with their heads firmly placed between the handler’s thumb and index finger. The 33-gauge Hamilton needle was held perpendicular to the mouse head and injected at a depth of approximately 3mm. The vector was slowly controlled and injected free handed into the lateral ventricles. At P1, a 33-gauge needle can easily penetrate the skull and there is no reflux of injectate as the ventricular system is sufficiently voluminous to accommodate the extra volume. Biodistribution of vector has been confirmed using this methodology9. For intravenous injections, P1 pups received intravenous injections of lentiviral or AAV vectors into the superficial temporal vein (20 μl) using a 33-gauge Hamilton needle. This procedure was performed free handed over approximately 5 seconds. Pups were returned as soon as possible to the dam and the nest undisturbed as much as possible. Animals were monitored daily for distress or discomfort such as the presence of a dull/ruffled coat, poor posture (hunched), pale or sunken eyes, a change in normal behaviour, reduced food or water intake, dehydration, any discomfort when handled, a reluctance to move, and any significant weight loss. In this cohort, we noted no significant signs of pain or discomfort following vector administration.
Validation and disease modelling induction of somatotransgenic bioimaging
Effects of carrier gas on hypoxia-inducible factor (HIF) induction
All mice received neonatal intracranial injection of lentivirus vector containing the HIF response element upstream of a luciferase-eGFP bicistronic cassette (LNT-HRE-Luc-eGFP) at P1. After weaning, age-matched mice were randomly allocated to two groups (N=11 for each group based on power calculations) for imaging at 0, 2, 4, 6 and 8 hours. Randomization was achieved by using Microsoft Excel to generate a random column of numbers against mouse numbers and then sorting the list of mice using the SORT function. The groups received isoflurane mixed with either 100% oxygen or air. The group receiving 100% oxygen exhibited consistently lower HIF activation than the group receiving air (21% oxygen); this was significantly different at 6 hours (P=0.0465, repeated measures ANOVA where one factor was with/without 21% oxygen and the repeated measure was time). See data availability section.
This data suggests that the carrier gas, itself, may influence physiological and pathological processes and supports the concept that conscious bioimaging may be preferable, where possible9.
Partial bile duct ligation in mice
All mice received neonatal intravenous injection of lentivirus vector containing the truncated GFAP promoter driving a luciferase-eGFP bicistronic cassette (LNT-GFAP-Luc-eGFP) at P1.
Mice were anaesthetized with 4% isoflurane at between 40–60 days after birth, shaved and disinfected around the surgical area, and a midline laparotomy (≈ 15 mm) performed just caudal to the sternum using aseptic surgery techniques. The median and left lobes of the liver were exteriorized and kept moist with sterile gauze. The bile duct was ligated with 6.0 silk suture to occlude outflow from the left and median lobes but not occlude bile outflow from the right and caudate lobes as described by Yang and colleagues13. The liver was returned to the abdomen and the wound closed in two stages using 6.0 silk suture; skin was closed using a subcuticular suture with buried knots (N=7). Sham operated mice received laparotomy only (N=5). Mice were recovered in a warmed chamber and monitored to make sure they were not under any unnecessary distress. Clinical record sheets were used to detail observations and post-operative treatments that were given. Pre- and immediate post-operative doses of topical bupivacaine hydrochloride (2 mg/kg) and subcutaneous 50 µl morphine (6.67 mg/ml) was administered for systemic post-operative analgesia14. Following surgery, mice were single housed and provided with moist food to aid consumption of food and prevent significant weight loss. Monitoring sheets recorded age, weight, disease and surgical status. Mice were monitored for approximately 1 hour post-surgery, and subsequently checked and weighed to monitor for overt signs of discomfort and significant weight loss. A loss of weight of more than 15% required humane euthanasia of the animal. In this cohort, the animals were not found to have negative post-operative outcomes, which is a significant improvement over performing full bile duct ligations which often result in pain, jaundice, and death to the animals. No animals within our cohort required euthanasia using partial bile duct ligation.
Whole-body bioluminescence imaging. Mice which underwent partial bile-duct ligations were anesthetized with isoflurane containing 100% oxygen, all other cohorts of mice described herein were imaged while conscious and free-moving. Mice received an intraperitoneal injection of 15 mg/mL of D-luciferin. Mice were imaged after 5 minutes using a cooled charged-coupled device camera (IVIS imaging machine) for between 1 second and 5 minutes. The regions of interest (ROI) were measured using Living Image Software and expressed as photons per second per centimeter squared per steradian (photons/second/cm2/sr). The imaging chambers used to image conscious mice for the intracranial injected mice had the following dimensions; 5 cm × 5 cm × 6 cm. The Perspex box which was used to image the conscious mice which had received an intravenous injection is described in detail in Karda et al.9. Where possible, BLI was carried out in adult reporter rodents on three consecutive days to establish a robust median baseline; subsequent data points were expressed as fold-change over this internal standard for each individual animal.
Euthanasia. At experiment conclusion, mice were culled using rapid cervical dislocation to induce rapid loss of consciousness. To reduce the possibility of operator error, the main concern for animal welfare using this technique, the procedure was performed by adequately trained personnel with a high degree of technical proficiency only. Where possible, cervical dislocation was performed while mice were still under non-recovery isoflurane anaesthetic. Death was assured by decapitation.
In consultation with a statistician, power calculations are performed based on previous data using the free GPower (v3.0.10) online software. Using the probability of finding an effect set at 80%, the probability of incorrectly rejecting the null hypothesis when it is true set at 0.05 (alpha), and using a two-sided test, a power calculation was performed in order to determine the sample size required for each group under investigation. Using our previous data involving the activation of the Smad2/3 binding element using the potent agonist, activin A, a standard deviation of 1.75 was used. The mean of group 1 (control) was set at 5.13, and the mean of group 2 was set at 7.8 (experimental). Based on these values, the effect size is expected to be large (1.52 fold change). An effect size greater than 0.8 is indicative of a large effect size, indicating that 93% of the control group will be below the mean of the experimental group. The experiment was sufficiently powered with a sample size of eight experimental and eight control animals to detect a change in the total amount of photonic output between two groups with a power of 80%. For in vivo data, repeated measurements following agonist administration were analyzed with repeated measures analysis of variance using GraphPad Prism v8.0.0 with a significance level of P <0.01. Area under curve data for experiments with two groups were analyzed by Student’s one-tailed t-test. For areas under curve derived from two or more groups, one-way analysis of variance (ANOVA) with Newman-Keuls post-hoc multiple comparison test was performed.
We have generated a comprehensive library of lentiviral TFAR vectors and are developing a similar library of AAV vectors (Table 1). These TFARs are available to any academic researcher on request. We also have developed a simple two-step protocol for shuttling de novo synthesized transcription factor binding sequences into our parental vectors.
TFAR design, synthesis, and sub-cloning
1. Derive minimal consensus binding sequence for the candidate transcription factor from the literature.
2. Design serial transcription factor binding sequence (TFBS) by interspersing 4-10 binding sequences with 10 random nucleotides using non-coding combinations of the 4 nucleotides.
3. De novo synthesize the resultant sequence flanked by BamHI (5’-) and EcoRI (3’-) restriction sites for directional cloning into pENTR-MP, a Gateway pENTR vector containing a minimal promoter sequence 5’-GGGCTATAAAAGGGGGTGGGGGCGCGTTCGTCCTCACTCTCTTCC-3’.
4. Clone TFBS into pENTR-MP using DH5α chemically competent cells to produce pENTR-TFAR.
5. Confirm clones by sequencing using the pENTR forward primer which binds approximately 82 bp from the BamHI cloning site. (ACTGATAGTGACCTGTTCGTTGC).
Cloning synthetic promoter into the lentiviral cassette.
1. Perform a single-step LR Clonase recombination reaction between the pENTR-MP vector containing the desired synthetic promoter with the LNT-GW-Luc-eGFP vector as per manufacturer’s standard instructions.
2. Transform 1 μl of the LR recombination reaction into One Shot™ Stbl3™ (Invitrogen) competent cells and grow cells overnight at 30°C on ampicillin-laden (100 μg/ml) LB-agar plates.
3. Select colonies and grow in a shaking incubator (280 rpm) overnight at 30°C in 5 mls of LB broth containing ampicillin (100 μg/ml).
4. Screen and select colonies for positive clones using the BamHI restriction enzyme and gel electrophoresis. Correct clones contain 4 fragments of 10,928, approximately 200-300 bp (depending on response element and minimal promoter cloned in), 81 bp, and 12 bp (not seen on gel). A plasmid that has not undergone recombination and is still the LNT-GW-Luc-eGFP parental backbone that does not contain the biosensing response element contains 5 fragments of the following sizes: 10, 928 bp, 857 bp, 702 bp, 228 bp, 12 bp (not seen on gel). Example gel provided as underlying data15.
5. Confirm clones by sequencing following the recombination reaction into the lentiviral backbone by using the cPPT forward primer (GTGCAGGGGAAAGAATAGTAG).
Cloning synthetic promoters into the AAV cassette.
1. As described above for cloning synthetic promoters into the lentiviral cassette, perform LR Clonase recombination reaction between pENTR-MP containing the synthetic promoter of choice with the AAV-GW-Luc-eGFP, as per manufacture’s protocol (see Note 1 for Clonase storage recommendations).
2. Transform 1 μl of the LR recombination reaction into One Shot™ Stbl3™ (Invitrogen) competent cells (see Note 2) and grow cells overnight at 30°C on ampicillin-laden (100 μg/ml) LB-agar plates.
3. Select colonies and grow in a shaking incubator (280 rpm) overnight at 30°C in 5 mls of LB broth containing ampicillin (100 μg/ml).
4. Screen colonies for positive clones using appropriate restriction enzyme depending on the desired synthetic promoter and then perform gel electrophoresis.
1. The LR Clonase enzyme mix is unstable even at -20°C for extended periods. Thus, it is recommended that small aliquots of 5 μl are made and preferably stored at -80°C to reduce the number of freeze/thaw cycles and retain as much enzyme activity.
2. One Shot™ Stbl3™ competent cells have been designed specifically for the propagation of unstable DNA sequence such as those found within the lentiviral backbone which contains direct repeats.
Generation of high-titer TFAR lentivirus.
1. Seed HEK293T cells (mycoplasma negative – see Note 3) at approximately 2x107 cells per T175 cm2 flask and incubate at 37°C, 5% CO2 overnight to achieve up to 90% confluence.
2. Mix lentiviral plasmids in Opti-MEM™ I reduced serum medium (7.5 mls / T175 flask) in the following quantities (per flask): 50 μg transfer vector containing transgene, 32.5 μg gag-pol packaging vector (pCMVΔR8.74) OR psPAX2 and 17.5 μg VSV-G envelope vector (pMD2.G).
3. In a separate tube, mix 1 μl 10 mM branched PEI in 7.5 mls Opti-MEM™ I reduced serum medium® (per flask). Incubate for 5 minutes.
4. Mix DNA and PEI solutions together in a 1:1 ratio (total of 15 mls per flask) and incubate at room temperature for 20 minutes to allow DNA:PEI complexes to form (see Note 4 for PEI preparation using branched PEI). This is hereon described as transfection media (Opti-MEM, DNA:PEI complex).
5. Remove media from cells in flask and add 15 mls of transfection media.
6. Incubate transfection media on cells in an incubator at 37°C / 5% CO2 for 3 hours.
7. Remove transfection media and replace with 15 mls complete media (DMEM + 10% FCS + 1% Pen/Strep).
8. After 24 hours, remove media and replenish cells with new culture medium.
9. After a further 48 hours, collect the virus-containing medium and filter through a 0.45 μm PVDF filter (see Note 5).
10. Subject virus-containing medium to overnight centrifugation (16-20 hours) at 5000g at 4°C.
11. Repeat harvest and overnight centrifugation for 72-hour post-transfection supernatant.
12. Resuspend the viral pellet in 50 μl Opti-MEM I reduced serum medium® (per T175 flask of cells used) and gently mix every 20 minutes for 1 hour at 4°C and cryopreserve at -80°C.
13. Lentiviral titer is obtained using a RETRO-TEK HIV-1 p24 antigen ELISA as per manufacturer’s protocol.
Generation of high-titer TFAR AAV.
1. Seed 6–7 × 106 HEK293T cells into 15cm plates so that they are 70-80% confluent and incubate at 37°C, 5% CO2 overnight.
2. Mix 10.5µg of the AAV transgene plasmid, 10.5µg of pDG8 plasmid expressing AAV2 Rep, AAV8 Cap gene and 31.5µg pHGT1 (Ad helper plasmid) to 1.5mls of Opti-MEM reduced serum medium. Add 758 μl of stock transfection agent PEI (2mg/ml) to Opti-MEM (1.5ml/dish), mix and leave at room temperature for 15-20 minutes.
3. Drop-wise, add 3 ml of transfection mix to each dish and gently distribute
4. After 24 hours remove media and replace with 15 mls of DMEM (2% fetal calf serum and 1% Pen/Strep antibiotic)
5. After 48 hours harvest the cells by scrapping the cells off the dish and pooling the media and cells to 50 ml.
6. Spin at 1500 rpm* for 5 minutes and pour supernatant into a receiver bottle and store at -20°C. *revolutions per minute (herein, on a centrifuge with size of rotor radius 17.4 cm).
7. Re-suspend pelleted cells in 1 ml TD lysis buffer (140mM NaCl, 5mM KCl, 0.7mM K2HPO4, 3.5mM MgCl2, 25mM Tris. pH7.5), pool and store in -80°C.
8. Freeze thaw pellet 5 times and add 25 ul/ml of 20% sodium deoxycholate.
9. Add 8 ul/40 mls of cells Benzonase and incubate at 37°C for 30 minutes. Spin at 4000rpm for 30 minutes at 18°C and filter using 0.45 um filters. Repeat this twice.
10. For supernatant, aliquot into 50 mls tubes and add 5ul of benzonase per 50mls of supernatant.
11. Add 100 ul of MgSO4 and incubate for 30 minutes at 37°C, after which spin at 4000 rpm for 30 minutes at 4°C. Filter using 0.45 um filters, repeat twice.
12. Run the cell pellet and supernatant through the HPLC machine using the POROS column16.
13. Add virus to dialysis cassette and place cassette into 1 L of PBS. Keep spinning overnight at room temperature on a slow rotating rotor.
14. Prime the membrane of the Amico Ultra tube by adding 5 mls PBS and centrifuge for 5minutes at 4000 rpm.
15. Remove PBS and place virus onto membrane. Spin at 4000 rpm for 5 minutes
16. Wash membrane with vector collected and add additional 100 ul PBS. Place vector into a 2 ml centrifugal tube with 0.22 um filter. Spin for 3 minutes at 13,000K on a benchtop centrifuge.
17. Remove filter and aliquot vector. Store at -80°C.
18. AAV vector was quantified using qPCR as per previously published protocol16, but briefly, samples were treated with DNase to release DNA. Serial dilutions were prepared to be used as standards. Both standards and samples were tested in triplicate. Cycling parameters were: 95°C for 10 minutes, followed by 40-45 cycles of 95°C for 15 seconds and 60°C for 45 seconds. Vector titers were calculated using the following formula: Titre (vc/mL) = C/5 × 1000 × D × 10 × 2 (See note 6).
3. Mycoplasma infections have the capacity to reduce lentiviral titers. Therefore, perform a mycoplasma test using PCR with the following primers: Forward (5’- gggagcaaacaggattagataccct - 3’) and Reverse (5’- tgcaccatctgtcactctgttaacctc -3’) with the following cycling parameters: 94°C for 30 seconds, 55°C for 30 seconds and 72°C for 30 seconds repeated for 30 cycles. A 1.5 - 2% gel will yield a 300 bp fragment on an agarose gel. Alternatively use a fluorescence-based method using a kit such as MycoAlert™ Mycoplasma Detection kit (Lonza).
4. To make up 10 mM branched PEI (MW ~25,000), in a fume hood, add 10 ml water to 10 ml PEI and vortex. Add 12 N HCL, 1 ml at a time, and vortex (this is approximately 10 ml HCL) then top up to 41.2 ml with water. Make sure that this final solution is at pH 7.0. Vortex and store in small aliquots at -80°C.
5. Both PES and PVDF filters are suitable but PVDF has been shown to be lower protein binding.
6. Where C is the copy number measured from the reaction, 5 is the volume of sample used, 1000 is the conversion factor from µl to ml, D is the dilution factor, 10 is the initial dilution of the AAV from the digestion (5 µl to 50 µl), and 2 takes into account the complementary strand that is not targeted by the primers within the reaction.
In vitro validation of HRE reporter constructs.
1. Plate cell lines at ~60% confluence and transduce with virus at an MOI of 10.
2. Amplify transduced cells for at least 72 hours before replating to allow the virus to integrate and express basal levels of the transgenes.
3. Add 250 µM CoCl2 in RPMI-1640 media to activate HRE activity for 12 hours followed by addition of 500 µl of RPMI-1640 media (see note 6).
4. At 24 hours post-activation, replace with complete media.
5. For luciferase activity, lyse approximately 5x105 cells in 300 μl luciferase lysis buffer (0.65% NP40, 10 mM Trizma® base (pH 8.0), 1 mM EDTA (pH 8.0), 150 mM NaCl)
6. Clarify the soluble fraction of the supernatant by centrifugation at 13,000xg for 1 minute.
7. Add 20 μl cell extract to 20 μl assay buffer (25 mM Trizma® base pH 7.8, 1 mM DTT, 1 mM EDTA, 1% Triton™ X-100, 8 mM MgCl2, 3 ml glycerol, 1.25 mM rATP, 0.5% BSA).
8. Inject D-luciferin substrate (Gold Biotechnology) into each well at a final concentration of 1.5 mM.
9. Measure luminescence output using a suitable microplate reader with luminescent capabilities.
10. Perform a protein assay to normalise relative photonic light units to total protein.
11. Only if a statistically significant response to agonist or antagonist was achieved, using appropriate statistical evaluation, would the TFAR be deemed suitable for in vivo experiments.
1. Administer concentrated lentiviral or AAV vector (5 μl total) by unilateral injection into the cerebral lateral ventricles using a 33-gauge Hamilton needle (see note 7).
2. Perform intravenous injections of lentiviral or AAV vectors (20 μl total) into the superficial temporal vein using a 33-gauge Hamilton needle.
1. As previously described9, anesthetize mice (see Note 8) with 4% isofluorane in 100% O2 (see Note 9). Inject 300 μl D-luciferin solution at a concentration of 15 mg/ml (a dose of approximately 150 mg/kg) into the intraperitoneal cavity (see Note 10).
2. Image the unconscious mice in the warmed lightproof detection chamber of the IVIS in vivo imaging system (see Note 11). Commence imaging 5 minutes after D-luciferin administration (t=0) (see Note 12). An overlay of the two images is generated using Living Image software to create a pseudo-colored image to depict luminescent intensities over each animal.
3. Define regions of interest (ROIs) manually using a standard area for each organ.
4. Prior to agonist-mediated activation or surgical induction of disease, each of the animals is administered with D-luciferin and imaged three times within 72 hours in order to ascertain a robust median baseline measurement of bioluminescent imaging which can subsequently be used to express all future data points as a fold-change over this baseline value.
5. The type of statistical test depends upon the nature of the biosensor and the kinetics of the response. Two possible approaches are; A) for each animal in the two experimental groups, obtain the area under the curve using the parallelogram method. Compare using a Student’s t-test if data is normally distributed. If mathematical transformations were not sufficient to satisfy parametric assumptions we suggest using a non-parametric test e.g. Mann-Whitney U-test. B) compare two or more experimental groups over time using analysis of variance (ANOVA) with repeated measures. If ANOVA shows a significant difference between groups, perform a post-hoc test (e.g. Tukey, Bonferroni or Sidak) to test which time points might be significantly different (see Note 13).
7. The 33-gauge Hamilton needle should be kept moist at the tip by placing a wet paper towel around the needle. This helps reducing the friction against the new-born mouse skin.
8. Mice can be injected without anesthesia. However, anesthesia reduces mobility and improves injection accuracy. Inhalation or injectable anesthetics are avoided as the nose cone impairs access to the injection sites (especially the superficial temporal vein).
9. Mice may also be anesthetized using air or air and a nitrous oxide mix. The choice of carrier gas may affect the chosen biosensor as well as firefly luciferase activity (since this is an oxygen-dependent reaction).
10. When administering the D-luciferin via intraperitoneal injection, make sure that the bladder or other internal organs are not penetrated by the needle. This can be achieved by “tenting” the skin for injection. Similarly, ensure penetration into the peritoneal cavity by watching for, and avoiding the formation of a subcutaneous bleb.
11. To achieve optimal luciferase expression from the mice, it is best to use white furred mice as the black-furred mice prevent the bioluminescence from penetrating through.
12. Waiting 5 minutes after D-luciferin administration permits time for entry of the D-luciferin into the bloodstream. It is worth performing a preliminary experiment to determine the kinetics for optimal bioluminescence for different cell and tissue targets. In addition, alternative routes of D-luciferin administration (e.g. intranasal) may be used.
13. It is good practice to perform a preliminary experiment to gauge the kinetics of the biosensor response and to identify timeframes of these responses in order to refine subsequent statistical tests. The statistical test and the time points of analysis should be decided before the experiment is performed, not afterwards.
Lentiviral delivery of vectors containing transcription factor binding elements upstream of a bicistronic reporter cassette offer a unique method with which to interrogate biological pathways. HRE is used as a biological sensor of hypoxia, important in cancer and ischemia models of disease. Here we present data validating our biosensing constructs in vitro (Figure 1) prior to in vivo (Figure 215) validation and experimentation.

HEK293T (A) and HeLa (B) cells were transduced with pLNT-HRE-Luc-eGFP and either activated (n=3) with 250 µM CoCl2 (red bars) or not activated (n=3) (grey bars) in triplicate for each of the time points (T = 12, 24, 48, and 72 hours). Data was analyzed using a two-way ANOVA with repeated measures with Bonferroni post-hoc test to correct for multiple testing. Bars represent mean ± SD with adjusted p values indicated above each timepoint (*=<0.05, **=<0.01, ***=<0.001, ****=<0.0001).
In vitro validation of the pLNT-HRE-Luc-eGFP construct. Prior to using the LNT-HRE-Luc-eGFP vector to interrogate signaling profiles in disease models in vivo, it was important to first validate this construct and establish its ability to be induced by agonists in vitro. CoCl2 is a chemical inducer of the hypoxia-inducible factor that mimics hypoxia. In the presence of CoCl2, a highly significant increase in luciferase-mediated bioluminescence fold-change (Δ bioluminescence) over non-activated cells was observed in vitro (Figure 215) prior to in vivo (Figure 315) validation and experimentation.
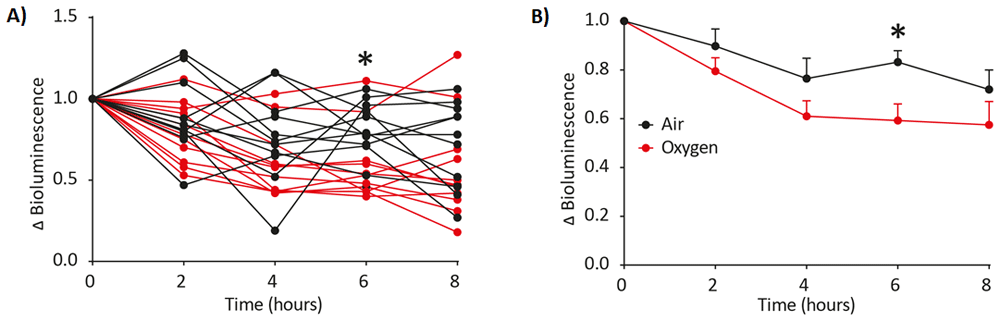
LNT-HRE-Luc-eGFP transduced animals were imaged at 0,2,4,6 and 8 hours in either 100% oxygen (n=11) (red) or air (n=11) (black) carrier gas. An increase in oxygen results in lower hypoxia-induced luciferase expression with data expressed as a fold change in bioluminescence (Δ bioluminescence) from time point 0. Δ Bioluminescence was plotted per individual animal (A) and as mean±SD (B). Statistical significance of > 0.05 is indicated by * (Sidak’s multiple comparison test, Graphpad Prism 8.0).
Response to oxygen content of carrier gasses during anesthesia. Following validation of the hypoxia response element in vitro, we chose to use it to ask whether in vivo HIF activity is modulated by the anesthetic carrier gas. 100% oxygen is frequently used as a carrier gas to ensure sufficient tissue oxygenation during anesthesia however, theoretically, it may downregulate HIF activation as this response element responds inversely to hypoxia, or the lack of oxygen. Mice underwent anesthesia with isoflurane using either air or 100% oxygen as the carrier gas and HRE signaling monitored at a series of time points using bioluminescence imaging. A change in bioluminescence (Δ bioluminescence) compared to time point 0 was noted over time with oxygen expectedly decreasing the expression of the HIF response element (Figure 3A-B15). This indicates the importance of selecting the correct carrier gas for anaesthetic induction when interrogating HIF-responsive signaling using luciferase expression and imaging.
Somatotransgenic bioimaging to assess GFAP activation in a model of cholestatic liver injury. The partial BDL model is an established model of cholestatic liver disease17. Currently, analysis of liver disease consists of terminal endpoint analysis of groups of animals at serial time points, and/or serial bleedings for analysis of serological biomarkers of liver function, particularly liver enzymes and total serum bile acids. We have previously studied signal transduction pathways of inflammation and WNT-signaling in this model14. Here, we asked whether expression of GFAP, a marker of activated hepatic stellate cells that are a major cell type involved in liver fibrosis, was altered following induction of liver injury. P1 mice received LNT-GFAP-Luc-eGFP, a lentiviral vector containing the luciferase-eGFP transgene driven by the bioresponsive glial fibrillary acidic protein (GFAP) response element. At 45 days of age mice were randomly allocated to two groups, one of which received partial bile duct ligation; controls received sham surgery without ligation of the bile duct (Figure 4A–B15). There was a consistent increase in bioluminescence 3 days after induction of the model versus sham controls. One particular mouse showed a remarkable, sustained increase in luciferase output following induction. This data illustrates the strength of continual non-invasive measurements, in that it is possible to identify consistent outliers, which would not be possible with terminal end-point analysis, due to a lack of data on expression kinetics.
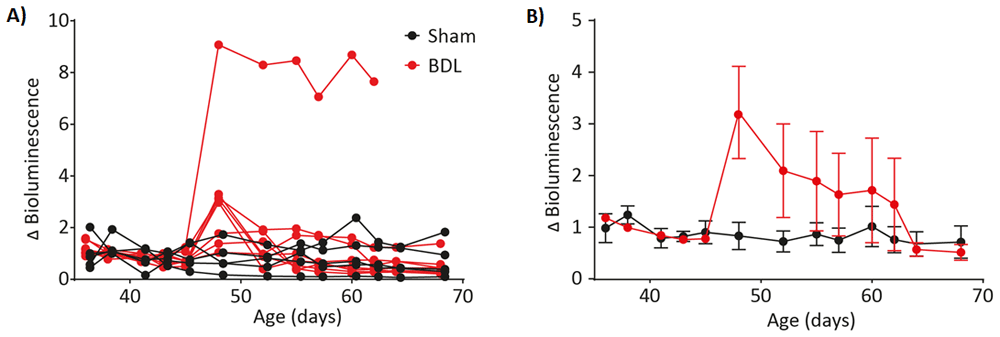
LNT-GFAP-Luc-eGFP-transduced animals were subject to partial bile duct ligation (n=7)(red) as a model of cholestatic liver injury. Another group of animals underwent sham surgery only (n=5)(black). Fold-change in bioluminescence (Δ bioluminescence) was calculated before and after model induction and plotted as individual animals (A) and means ± SD (B).
The development of the described methodologies has a broad scope of applicability for monitoring of disease progression, induction, and therapeutic intervention. The generation of biosensing, reporter animals that can undergo longitudinal assessment of transcription factor activity has significant implications for the number of animals currently being utilized within both academic and industry research and development. This novel platform of somatotransgenic bioimaging is also capable of generating longitudinal datasets from small cohorts of animals. F0 cohorts based on appropriate power calculations can be administered with TFAR vector and assayed almost immediately, depending on the experiment, after drug administration or disease state induction. In many instances, animals can be reused for further follow-up studies without any due stress or requirement for serial anesthesia. This approach results in a massive reduction in animals required for a predicted outcome and a clear decrease in procedures stressful to animals.
Open Science Framework: Non-invasive somatotransgenic bioimaging in living animals. https://doi.org/10.17605/OSF.IO/XF7EZ15
This project contains the following underlying data:
1. 293T – HRE – CoCl2 – Luciferase assay calculations.csv - (in vitro luciferase assay using the LNT-HRE-Luc-eGFP and cobalt chloride agonist in HEK293T cells).
2. HeLa – HRE – CoCl2 – Luciferase assay calculations.csv - (in vitro luciferase assay using the LNT-HRE-Luc-eGFP and cobalt chloride agonist in HeLa cells).
3. BDL NC3Rs raw data.csv - (In vivo raw data for GFAP activation using bile duct ligations (BDL)).
4. HRE NC3Rs raw data.csv - (In vivo raw data for HRE activation using the model of hypoxia).
5. pLNT-GW-Luc-eGFP.dna – (plasmid map of the lentiviral construct containing the Gateway cassette upstream of luciferase and eGFP).
6. pLNT-HRE-Luc-eGFP.dna – (plasmid map of the lentiviral construct containing the HIF response element (HRE) upstream of luciferase and eGFP following recombination).
7. Gel image of LNT-GW-Luc-eGFP before recombination.tif – (BamHI digest of LNT-GW-Luc-eGFP prior to recombination reaction).
8. BamHI digest of pLNT-HRE, WNT, PI3, HNF4, Gli-Luc-eGFP clones.tif – (BamHI digest of LNT-HRE, WNT, PI3, HNF4, Gli-Luc-eGFP response elements following recombination reaction).
9. pLNT-HRE-Luc-eGFP sequencing to confirm HRE.abi – (sequencing of LNT-HRE-JDG and alignment to theoretical sequence of LNT-HRE-JDG).
Data are available under the terms of the Creative Commons Zero "No rights reserved" data waiver (CC0 1.0 Public domain dedication).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)