Keywords
Spatial organization, single cell RNA-seq, Drosophila embryo, clustering, DREAM challenge
Spatial organization, single cell RNA-seq, Drosophila embryo, clustering, DREAM challenge
The development of single cell RNA sequencing (scRNA-seq) has provided a powerful solution for building a global transcription landscape of all cell types in tissues, finding new cell types, cell lineage tracing, spatial reconstruction, and combining with other omics1–4. The single cell is originally made from dissociated tissues without spatial information, and spatial gene expression pattern is unknown. In situ hybridization (ISH) and its variants can detect the spatial information of mRNA transcripts and produce gene expression reference atlas. Using enough marker genes, users can reconstruct the spatial position of single cell RNA-seq data by combing the ISH reference atlas3–7. Some works have also combined sequential fluorescence in situ hybridization (seqFISH) and multiplexed error robust fluorescence in situ hybridization (MERFISH) with scRNA-seq data to map cell types to the reference atlas8–10.
Recent methods have successfully mapped cells from scRNA-seq data to the spatial positions using dozens of landmark genes3–7. Nikos et al. developed a DistMap method for mapping the ~1300 Drosophila embryo cells into ~3000 bins in the spatial position using 84 marker genes3. Satija4 et al. mapped 851 cells of zebrafish embryo into 64 bins in spatial embryo using 47 genes. Kaia et al. computed the correspondence score and mapped 139 cells into a Platynereis dumerilii brain using a set of 98 genes5. Andreas et al. reconstructed spatial single enterocytes along the villus axis in 1-D space using 50 bottom and top landmark genes for 1383 cells6. Mor et al. proposed novoSpaRc for spatial mapping of the scRNA-seq cells into an existing reference atlas to infer spatial gene expression7. In these methods, the dimension and resolution of the spatial region, as well as the number of marker genes, are key factors to affect the recovery of the spatial position.
The DREAM Single Cell Transcriptomics Challenge aims to develop new algorithms to find embryo spatial pattern. Participating teams are asked to predict the positions in the embryo of 1297 cells using the expression pattern from 60 (sub challenge 1), 40 (sub challenge 2), and 20 (sub challenge 3) driver genes from in situ hybridization data. The challenge is different from the published methods as it endeavors to use less marker genes to infer the spatial locations of cells.
In this article, we introduce an unsupervised learning approach for the three challenges, and validate the results using the “gold standard” method derived from DistMap which uses 84 genes. The paper is organized as follows: Methods, we briefly describe the solutions for all three sub-challenges; Results, we present results of three sub-challenges on the data of the DREAM Single Cell Transcriptomics Challenge; finally, we discuss our results and summarize our work.
The dataset is from Drosophila embryos. The scRNA-seq dataset is from ~1000 handpicked stage 6 fly embryos using Drop-seq11. It contains both raw and normalized UMI counts with 1297 cells and 8924 genes per cell. A total of 84 driver genes are used. In situ hybridization expression patterns are from the Berkeley Drosophila Transcription Network Project (BDNTP). The BDNTP reference atlas are binarized. The bin number of one half of the embryo is 3039. The spatial coordinates of these bins are also specified. The dataset files can be downloaded from the DREAM Single Cell Transcriptomics Challenge after registration with Synapse free of charge (https://www.synapse.org/#!Synapse:syn16782360).
We directly use the normalized scRNA-seq data, the in situ matrix and the geometry of the embryo. The gene names “E.spl.m5.HLH” and “Blimp.1” are replaced by “E(spl)m5-HLH” and “Blimp-1”.
We use a hierarchical clustering method to select 60, 40, and 20 driver genes from the 84 genes based on the normalized scRNA-seq data.
Based on the belief that the scRNA-seq gene expression pattern is similar to the driver genes’ pattern, we propose to select the essential driver genes based on the information provided by scRNA-seq data. Namely, if two genes have high correlation in the scRNA-seq data, we assume the same pattern happens in the in situ matrix. Therefore, we choose only one of them without losing too much of the information. To find the correlated genes, we perform hierarchical clustering on the normalized scRNA-seq data to separate all 84 genes into 60 clusters (with the Euclidean distance and the Mcquitty linkage). The Mcquitty linkage gives more weights for objects in small clusters than those in large clusters in calculating the distance between two clusters. Thus, it is suitable for situations with many small clusters. Since the numbers of clusters are fairly large in the sub-challenge 1 and 2, we opt to use the Mcquitty linkage for distance calculation. In sub-challenge 3, since the total number of clusters is shrunk to 20, which is smaller than sub-challenge 1 and 2, we choose to use the ward linkage in the hierarchical clustering part to obtain larger-sized clusters from the data. After this step, the gene selection process remains the same as sub-challenge 1 and 2.
After getting the clusters, we pick the most representative gene of each cluster by calculating the distance between each member gene and the cluster center based on the Euclidean distance and selecting the closest one.
We perform binarization on the normalized scRNA-seq data for the selected genes based on the “binarizeSingleCellData()” function in DistMap (https://github.com/rajewsky-lab/distmap). The details of binarization is as follows: for each quantile threshold, we perform binarization on the scRNA-seq data for each gene. If the gene expression value is larger than the quantile gene expression value, it will be set as 1, otherwise it is 0. Then we compute the difference between the correlation matrix of binarized scRNA-seq data and the correlation matrix of in situ matrix based on the root-mean-square error. Last, we select the quantile threshold which has the smallest difference to perform binarization for scRNA-seq data.
Given the binarized scRNA-seq data and in situ matrix, we calculate the probability matrix between cells and bins based on the selected driver genes. Here, we assume the selected driver gene number as ng. The probability pij of a cell ci (i∈[1,1297]) originating from the bin bj (j∈[1,3039]) can be expressed as follows.
is the number of the same gene expression value (0 or 1) in the two binarized vectors of the scRNA-seq data and the in situ matrix for cell ci and bin bj.
The probability of a cell originating from a bin is determined by the gene expression in the bin and cell. More genes can improve the prediction of cell position. The bins with the higher probability are possibly the potential cell position. For sub-challenges 1–3, we follow the same process shown in Figure 1. To make the results more stable, we select enough bins (see below) based on the probability values. Then we use clustering to determine a more stable cell position.
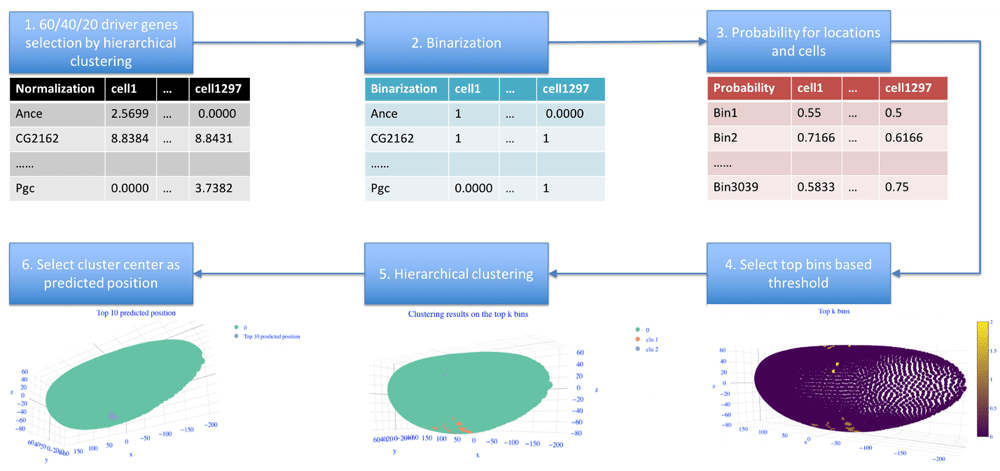
To select the potential bins for predicting cell position, we check the distribution of the maximum values in the probability matrix for the bins (Figure 2). Then we use the third quartile of probability values to select the top bins in sub challenge 1 when using 60 driver genes. We use the first quartile of probability values in sub challenge 2 when using 40 driver genes. And we use all bins in sub challenge 3 when using 20 driver genes. If the number of selected bins is 0, the bin which has the maximum probability will be the predicted position. If the number of selected bins is larger than 100, then only the top 100 bins will be kept based on the probability.
To check the effect of threshold on the prediction results, we test our method under different thresholds as shown in Figure 4 (b)–(d).
For high probability bins, we need to perform clustering to select the cluster which has the maximum sum of probability as cell position. Here, we use hierarchical clustering on the selected bins. The cluster number is determined by the silhouette score, which measures the average distance of a point to other points in its cluster compared to the smallest average distance to other clusters. The silhouette score ranges from -1 to +1. The higher the silhouette score, the closer the point is closer to its own cluster and the farer it is away from other clusters.
We use the average silhouette score across all points to select the clustering number. We use NbClust package12 to perform hierarchical clustering with the “centroid” method. Based on the silhouette score, we obtain the best clustering number. Then we compare the sums of probabilities of all clusters, and select the cluster which has the maximum sum of probabilities. We use the selected cluster center as a reference point to select 10 nearest bins as the top 10 most possible cell positions.
To evaluate the performance of our method, we use the three performance scores in the DREAM challenge (https://www.synapse.org/#!Synapse:syn17091286, https://github.com/dream-sctc/Scoring/blob/master/dream_scoring_clean.R). The first scoring metric is the primary score to estimate the precision of the assignment for the single cells. The second scoring metric is the average of the relative assignment metrics over all the single cells which is used when the first scores are equal for two methods. The third scoring metric is comparing prediction of gene patterns.
Ambiguous cells: If the predicted top 1 and top 2 positions are the same in the DistMap results, the prediction position will be ambiguous, and the cell will not be computed in the score 1–3. In this challenge, the number of ambiguous cells derived from DistMap are 287.
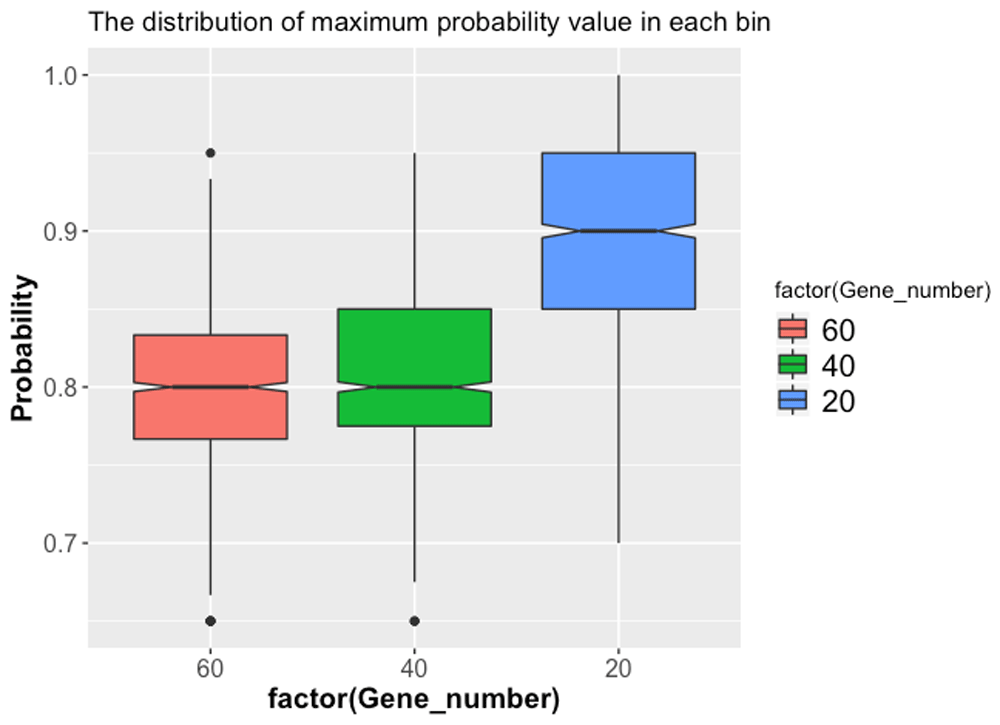
We calculated the sums of gene expression values in the in situ matrix for all selected driver genes for all 3039 bins. As Figure 3(a) shows, each bin has at least one gene expressed in the in situ matrix. It suggests that our selected driver genes can cover all bins. As the gene number decreases, the frequency of gene expression in each bin decreases. We also compared the overlapped genes in Figure 3(b). Among the 40 driver genes of the sub challenge 2, only one driver gene is not in the selected 60 driver genes of the sub challenge 1. Similarly, only 2 driver genes of the sub challenge 3 are not in the selected 60 driver genes. It suggests our method is consistent in selecting different number of driver genes.
(a) The frequency of gene expression in each bin in the in situ matrix in different selected driver genes scenarios. (b) The overlaps among the selected genes from the three sub challenges.
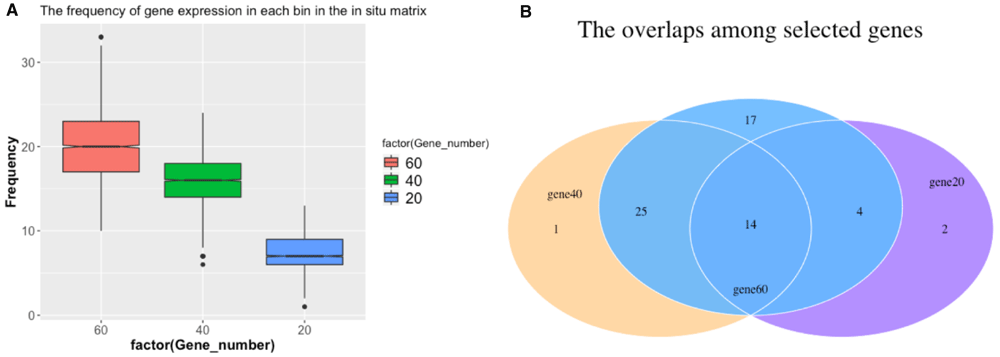
We used the score 1–3 to evaluate our method under the different selected driver genes scenarios. Figure 4(a) shows the scores of our submitted results for the sub-challenge 1, 2, 3. The blue bar is the score for the gold standard method using 84 driver genes from DistMap. For score 1, our method is close to the gold standard in sub challenge 1 when using 60 driver genes. The results of our method in sub challenge 2, 3 shows a larger difference. For score 2, our method shows high scores when using 60 and 40 driver genes. Score 2 is the average relative precision for all cells. It suggests that our method is robust for predicting the right position. The score 3 shows a small difference in our method when using 60 and 40 driver genes. Figure 4(b)-(d) shows the consistency of the score 1–3 over a range of thresholds in the different numbers of driver genes scenarios. Hence, Figure 4 shows that our method can obtain a close performance to the gold standard when using 60 driver genes.
(a) Comparing the score 1, 2, 3 for sub challenge 1 (60 driver genes), 2 (40 driver genes) and 3 (20 driver genes) with the gold standard (84 driver genes). (b–d) Comparing the score 1, 2, 3 using different thresholds for (b) sub challenge 1; (c) sub challenge 2; (d) sub challenge 3. (The numbers are rounded for visualization.)
As shown in Figure 5, the spatial gene expression prediction accuracy is represented by MCC correlation between the predicted cell position in the in situ matrix and the binarized scRNA-seq data for each driver gene. Score 3 is based on the MCC correlation for each driver gene used in each sub challenge. Corresponding to Figure 4(a), the MCC between the DistMap (84) and our method (60 or 40) for each driver gene in sub challenges 1 and 2 are very close. In sub challenge 3, the MCC of gene “dpn”, “erm”, “ftz”, “h” from our method are much lower than DistMap. It is consistent to the lower score 3 in sub challenge 3.
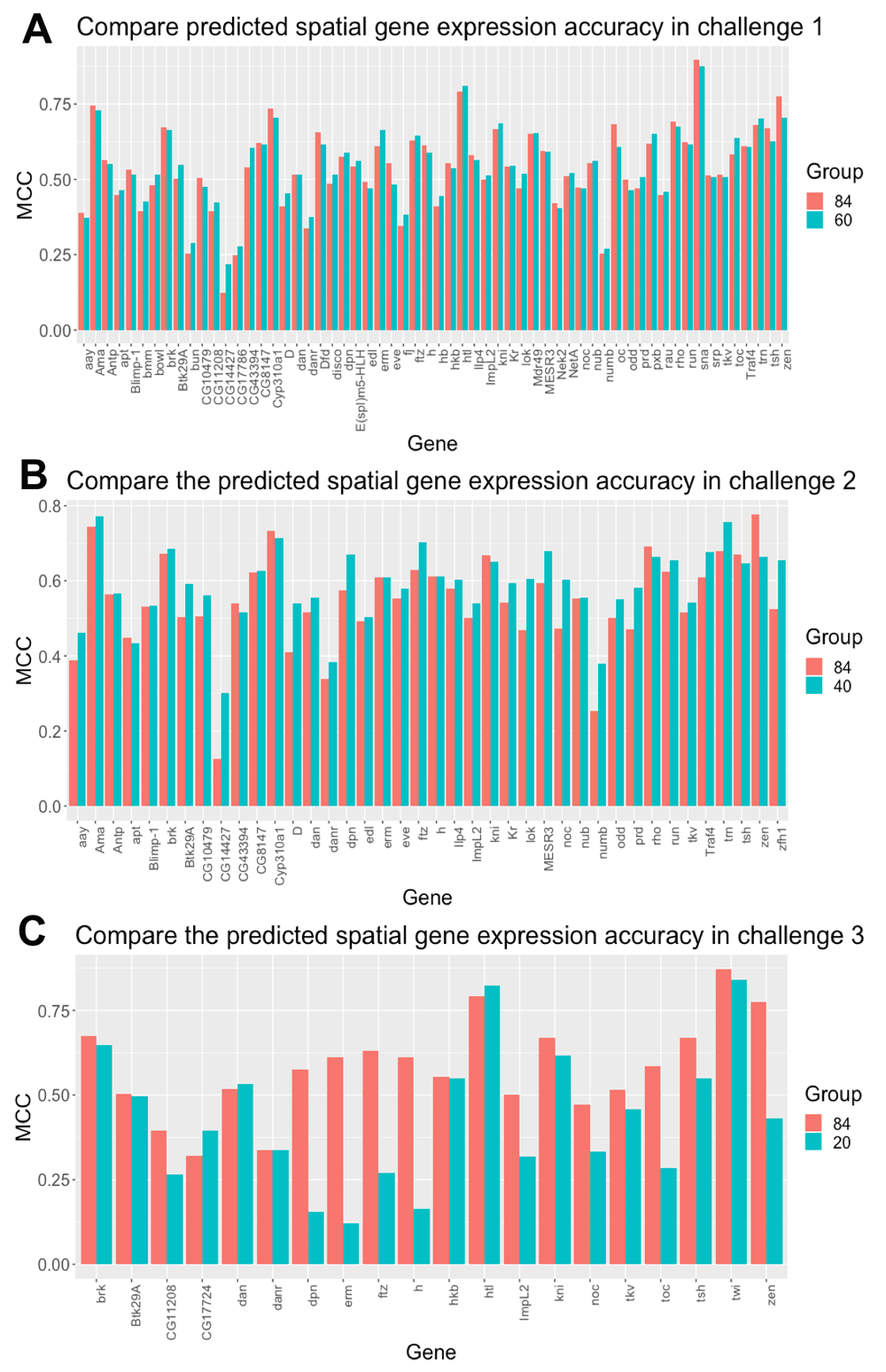
Comparing the spatial gene expression between DistMap (84) with our method using (a) 60 genes in sub challenge 1; (b) 40 genes in sub challenge 2; (c) 20 genes in sub challenge 3.
We described our method and its performance using 60, 40 and 20 driver genes by comparing with the gold standard (DistMap results). In sub challenge 1, our results shows a close performance to the gold standard. In sub challenges 2 and 3, when using 40 and 20 driver genes, the score 1 decreases and score 3 is still close to the gold standard. It suggests our method can predict cell positions using 60 genes and predict gene expression patterns using less genes. We tested the threshold for selecting top bins (Figure 4(b)–(d)): the results suggest that our method can achieve even better results when using the maximum threshold for sub challenges 1, 2, and 3.
The dataset associated with the DREAM Single Cell Transcriptomics Challenge is available for registered participants at https://www.synapse.org/#!Synapse:syn16782360 and https://www.synapse.org/#!Synapse:syn18632189. Due to sharing protocol of Synapse, users should register in Synapse (free of charge; https://www.synapse.org/) using their email address, and agree to the dataset conditions of use. Once registered, users can download the files.
Synapse: SCTC Challenge zho_team Submission. Code and results underlying this article, https://doi.org/10.7303/syn1705643513.
Source code implementation for the method presented in this article and used in the DREAM Single Cell Transcriptomics Challenge is available from: https://github.com/ouyang-lab/SCTC-Challenge-zho_team. Scoring scripts are available from: https://github.com/dream-sctc/Scoring/blob/master/dream_scoring_clean.R.
Archived source code as at time of publication: https://doi.org/10.5281/zenodo.359253214
License: GLP 3.0
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)