Keywords
Diatraea saccharalis, sugarcane borer, draft genome, assembly, Lepidoptera
This article is included in the Genomics and Genetics gateway.
Diatraea saccharalis, sugarcane borer, draft genome, assembly, Lepidoptera
Diatraea saccharalis (Fabricius), commonly known as the sugarcane borer, is an important moth pest of the family Crambidae (Lepidoptera; Crambidae) that is distributed throughout the Americas, including South America, the Caribbean, Central America, and the southeastern United States (Box, 1931; CAB International, 1989; Dyar & Heinrich, 1927). D. saccharalis host plants include important crops such as sugarcane (Saccharum spp.), sorghum (Sorghum bicolor), wheat (Triticum spp.), maize (Zea mays L.), and rice (Oryza sativa L.) (Myers, 1932; Rodríguez-del-Bosque et al., 1988; Roe, 1981). The damage caused by the D. saccharalis larvae in young plants produce a characteristic damage, that is the appearance in series of holes across the leaves that are still rolled up, and posteriorly begin to feed off the apical meristem that is killed, resulting in a condition known as "dead heart". In the developed plants, the larvae, after hatching, start to feed on the leaves and migrate to the sheath region where they are protected, and feed on their sheath and start to scrape the stalk. As the borer develops, generally third instar and older, it feeds almost exclusively within tunnels in stalks (Flynn et al., 1984). As a result of the borer behavior, the physical strength of the mature plant stalk is reduced, decreasing the plant biomass and sugar content in more developed crops, and have an increased susceptibility to plant pathogens due to the holes made by the larvae (Cruz, 2007; Wilson et al., 2017). In maize fields, a new behavior of the D. saccharalis was observed, where the larva may burrow into corn ears (Rodriguez-del-Bosque et al., 1990).
Although the sugarcane borer has a wide distribution across the Western Hemisphere (Dyar & Heinrich, 1927), it is treated as a single species and its identification relies on morphological characteristics despite the high variability shown in these traits between populations (Dyar & Heinrich, 1927; Francischini et al., 2017; Pashley et al., 1990). Several studies have developed molecular markers to provide a solid differentiation from other species of the same genus (Bravo et al., 2008; Francischini et al., 2017; Joyce et al., 2016; Pavinato et al., 2013; Pavinato et al., 2017) and to investigate the genetic variation among populations of D. saccharalis (Joyce et al., 2014; Joyce et al., 2016; Silva-Brandão et al., 2015). Geographical comparisons using individuals from the southeastern United States and Argentina have also suggested that the species might be treated as a cryptic species complex due to the significant genetic divergence among lineages (Fogliata et al., 2019; Joyce et al., 2014). The high genetic variation and a wide plant host range have also supported the success of this pest in colonizing regions with different environmental conditions, and human migration may likely be a major factor that contributed to its spread throughout the Americas (Francischini et al., 2019). Furthermore, the great increase of genetic diversity of the sugarcane borer in Brazil coincides with the expansion of agricultural production of sugarcane and maize crops in the Brazilian landscape (Pavinato et al., 2018). This makes D. saccharalis an interesting organism to study a wide range of questions including ecological adaptation and evolutionary dynamics. However, such studies require a significant number of molecular resources which are currently missing. A reference mitochondrial genome (Li et al., 2011) and a transcriptome assembly have been described (Merlin & Cônsoli, 2019); however, a well-assembled reference genome of D. saccharalis is not yet available.
Across the approximate 170,000 lepidopteran species, there are about 80 published genomes to date (Triant et al., 2018), however only a few of them have scaffolds assigned to the chromosome level [Bombyx mori (Kawamoto et al., 2019), Heliconius melpomene (Heliconius Genome Consortium, 2012), Plutella xylostella (You et al., 2013), Melitaea cinxia (Ahola et al., 2014), Trichoplusia ni (Chen et al., 2019), and Cydia pomonella (Wan et al., 2019)]. Recently, a few de novo genome assemblies of moth species have been released, such as Spodoptera frugiperda (Gouin et al., 2017; Kakumani et al., 2014), Thaumetopoea pityocampa (Gschloessl et al., 2018b) and Achroia grisella (Koseva et al., 2019). Due to the complexity of insect genomes, the draft assemblies have poor continuity and contain many gaps, which may result in a loss of a large number of genomic regions which relate to biological features of the species (Li et al., 2019). The lack of genetic information of this clade makes it difficult to understand some evolutionary processes such as those underlying the adaptation to plant defense systems under different environments (Davey et al., 2016). The rise of next generation sequencing technologies and the improvement in assembly algorithms have been providing more resources to assemble large and more complex genomes of non-model organisms at a lower cost (Wachi et al., 2018). Nevertheless, many challenges still remain when selecting the best assembly software and setting the best parameters in order to get the most accurate genome assembly.
In this study, we report the first genome sequence of D. saccharalis which was assembled using paired-end (PE) short reads generated by the Illumina HiSeq platform with high sequencing depth coverage. This draft genome assembly reveals important aspects of the genetics for this species such as genome size and heterozygosity levels. In addition, we identified hundreds of protein-coding gene models using a homology-based approach using B. mori protein information and investigated the functional annotation of these proteins. However, the difficulties in assembling long tandem-repeat regions using a short-read technology also resulted in fragmented sequences within the assembly. These data provide a vastly improved genome that will serve as a valuable resource for further genomic studies of the species in order to develop more effective approaches for the management of this pest.
D. saccharalis moths used for the DNA extraction were from an inbred colony which originated in Houma, Louisiana, and was reared on an artificial diet (Southland Products, Lake Village Arkansas). The insects were raised for about 70 generations, with new insects occasionally added to the colony. The adult moths were placed in 100% ethanol and shipped to the lab for DNA extraction. Genomic DNA was isolated from thorax and leg tissues derived from a single adult male specimen. The tissues were ground up to become a fine powder in a 10 mM Tris-HCl (Sigma-Aldrich, USA), 400 mM NaCl (Fisher Scientific, USA) and 2 mM EDTA (Fisher Scientific, USA) solution with the addition of RNase A (New England BioLabs, USA), and the high molecular weight DNA was precipitated using 120 µl 5M NaCl (Fisher Scientific, USA) (Miller et al., 1988). Prior to sequencing, we assessed the DNA concentration (620 ng) using a NanoDrop ND-1000 spectrometer (v3.8.1, Thermo Scientific, USA) and fragment size (18.2 kb) in a 0.7% agarose gel (Fisher Scientific, USA). A PCR-free shotgun strategy was used to prepare the genomic library with insert sizes ranging from 200 to 500 bp. Raw sequence data from paired-end libraries with read lengths of 2x250 were generated by an Illumina HiSeq 2500 sequencer at the University of Illinois at Urbana-Champaign.
Prior to the assembly, the quality of raw read data from sequencing was assessed using FastQC v.0.11.8 (Andrews, 2010) and preprocessed to remove adapter sequences, base-calling duplicates, as well as the elimination of low-quality reads (Phred Score < 30) using the Trimmomatic tool v.0.39 (Bolger et al., 2014). Based on assembly statistics, an optimal setup was found using ~40 million reads randomly selected which corresponds to a coverage close to 60X.
Genome size and heterozygosity rate estimations were made using a k-mer frequency distribution approach (Li & Waterman, 2003). Initially, the occurrence of unique 21-mers (where k=21) was counted and a distribution histogram generated using Jellyfish software v.2.0 (Marçais & Kingsford, 2011). The results were plotted using the Genome Scope tool v.0.1 (Vurture et al., 2017) with recommended parameters.
The genome assembly was constructed using SPAdes v. 3.11.1 (Bankevich et al., 2012). Briefly, the assembly process starts with the selection of multiple k-mer sizes (k = 21,33,55,77,99, and 127), followed by the construction of individual De Bruijn graphs for each size of k. Afterward, graphs were combined to calculate the distances between the k-mers and to map the edges of the final assembly graph. A set of contiguous DNA sequences, defined as contigs, were generated as output from SPAdes. Scaffolds were produced by combining contigs using read pair information and gap filling.
Following the genome assembly, we used an established MAKER v.2 pipeline (Holt & Yandell, 2011) to predict gene structure within the D. saccharalis scaffolds longer than 50 kb. First, repeat sequences were masked using RepeatMasker v.4.1.0 (Smit et al., 2013–2015), based on known repeat elements in RepBase (Bao et al., 2015). A total of 22,510 protein sequences from the lepidopteran silkmoth B. mori reference assembly (Duan et al., 2010) were used for the homology-based prediction approach. A Python script (Santos, 2020) was made to filter out gene models having low-similarity scores (Aligned identity < 50%) when compared to the reference protein.
The predicted protein dataset was imported into Blast2GO Basic software v.5.2.5 (Götz et al., 2008) for functional annotation analysis. Blast alignments were made using the NR database (NCBI, 1988) with Taxonomy Filter parameter set to “moths” and E-value threshold ≥ 1.0e-03. GO annotations were made in accordance with the Blast2GO protocols.
To access the fraction of the genomic sequences from D. saccharalis that corresponded to repetitive elements we first identified repeated families using RepeatModeler v.1.0.11 (Smit & Hubley, 2010) with the parameter “-engine ncbi” set. The custom library generated in this step was then used to mask the genome on RepeatMasker v.4.1.0 (Smit et al., 2013) with default parameters. The same procedures were also applied to the B. mori reference genome (Kawamoto et al., 2019) to compare the proportion of repeat content between these two species and verify the accuracy of this approach.
To assess the accuracy of the genomic sequences, we mapped the total set of Illumina raw paired-end reads to the assembled scaffolds using the BBMap software v.38.36 (Bushnell, 2014) to analyze the accordance rate between the primary dataset and the final assembly. Subsequently, we searched for the presence and completeness of highly conserved single copy orthologs from the “Insecta” database using BUSCO v.3.0 (Simão et al., 2015). The results were then compared to B. mori reference assembly results.
The genome of D. saccharalis was sequenced using the Illumina HiSeq 2500 system with paired-end 2x250 nt reads. We obtained over 306 Megabase pairs (Mb) of paired-end sequence data, representing 212X average genome coverage.
The haploid genome size estimate was made using the abundance of unique k-mers (k = 21), which corresponds to the second peak, highlighted by the dotted line in the plot (Figure 1). GenomeScope estimated a haploid genome size of 359 Mb. Additionally, this analysis revealed 0.69% of variation across the genomic sequences, indicating that the D. saccharalis genome of the selected individual had low heterozygosity properties, most likely due to the inbred nature of the colony used in this experiment.
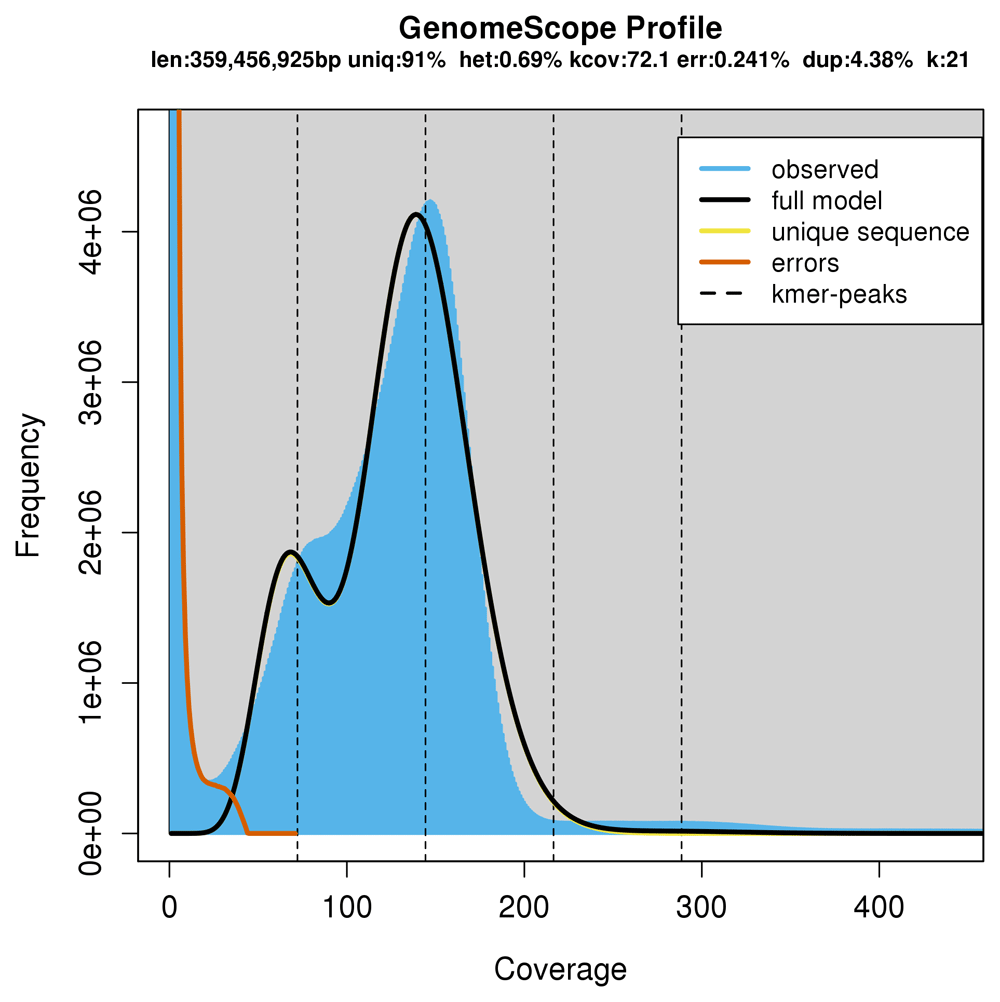
The first peak corresponds to the heterozygous and the second, the homozygous peak. Estimate of the heterozygous portion is 0.69%. Values evidenced in the subtitle correspond to the inferred total genome length (len), genome unique length percent (uniq), overall heterozygosity rate (het), mean k-mer coverage for heterozygous bases (kcov), read error rate (err), average rate of read duplications (dup) and k-mer size (k).
An optimal coverage size of input data was defined as 60X, corresponding to 46 million randomly selected reads obtained by a Python script (Santos, 2020). The assembly of the reduced data set using SPAdes resulted in 50,460 scaffolds larger than 1 kb having a cumulative length of 453 Mb, and a scaffold N50, an indication of assembly contiguity, of 16.3 kb. Additionally, the genomic GC content corresponds to 33.75% of the bases. Summary statistics of the assembly are described in Table 1.
The alignment of raw input short-read data resulted in a total of 96.40% of the reads mapped to the assembly, of which 89.64% were properly paired to the scaffolds. To access the completeness of our D. saccharalis genome assembly, we used BUSCO (Simão et al., 2015) and a set of 1,658 conserved, single-copy Insecta genes. We found that our assembly was highly complete, with 87.1% (1,445 out of 1,658) of these BUSCO genes being present in the genomic sequences (86.5% single copy, 0.6% duplicated genes). The B. mori reference genome contains 98.5% complete BUSCO genes (Figure 2).
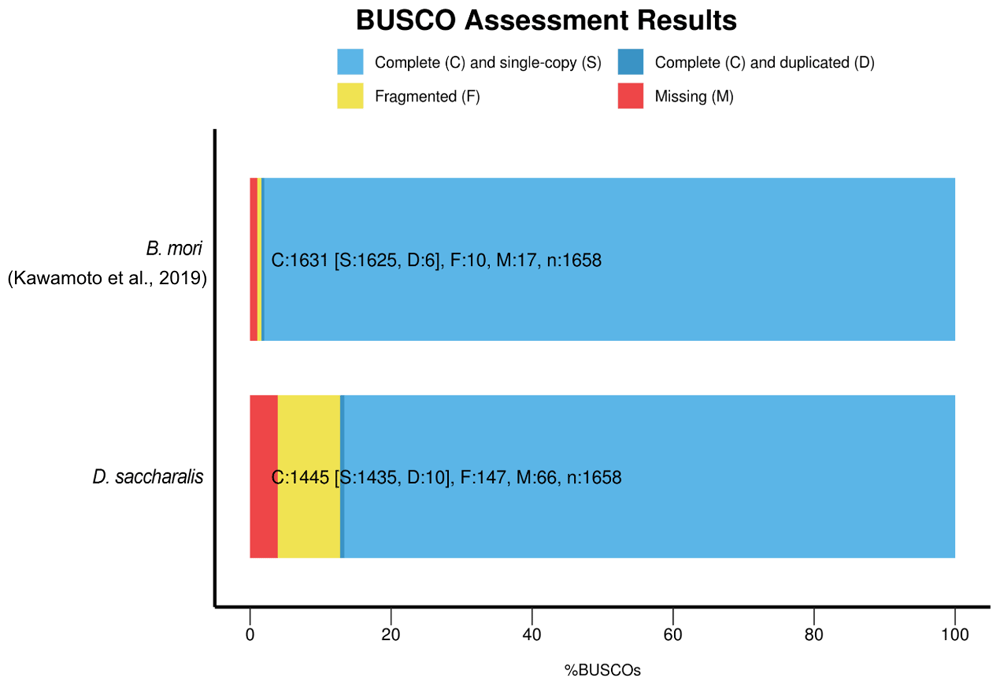
Bars show the proportions of conserved single-copy genes found in each assembly as a proportion of the total gene set (n).
Our analysis revealed that approximately 186 Mb of the D. saccharalis genome were identified to be various nucleotide repeat elements of which represent over 41% of the assembled sequences (Table 2). Although the majority of these elements were unclassified (35.44%), the repeats are composed by 14.8 Mb of long interspersed nuclear elements (LINEs), 4.5 Mb of simple repeats, 3.9 Mb of DNA transposons, as well as retrotransposons and low complexity elements. The consistency of our findings to the sugarcane borer assembly was assessed by analyzing the repeat content present in B. mori chromosomes through the same pipeline. B. mori assembly has shown a similar pattern of high repeat genomic content, having 46.32% of its genome composed of repeated elements. Moreover, the number of unclassified elements also compose a significant part of the repeats (112.6 Mb), followed by LINEs (70 Mb), DNA transposons (11.2 Mb), retrotransposons (4.8 Mb) and simple repeats (6.1 Mb).
| Repeat type1 | D. saccharalis | B. mori | ||
|---|---|---|---|---|
| Length (Mb) | % genome | Length (Mb) | % genome | |
| LINEs | 14.88 | 3.28 | 70.66 | 15.87 |
| LTR | 0.36 | 0.08 | 4.83 | 0.97 |
| DNA Transposons | 3.9 | 0.86 | 11.22 | 2.52 |
| Unclassified | 160.61 | 35.44 | 112.69 | 25.32 |
| Simple Repeats | 4.5 | 0.99 | 6.16 | 1.39 |
| Low Complexity | 0.7 | 0.16 | 0.56 | 0.13 |
| Total | 186.66 | 41.18 | 206.16 | 46.32 |
A total of 548 D. saccharalis genomic sequences longer than 50 kb were used in the MAKER homology-based gene prediction pipeline along with a library of repeated elements generated using RepeatModeler v.1.0.11 and the B. mori protein dataset as reference for building the models. The analysis resulted in a total of 1,394 predicted gene models among the D. saccharalis scaffolds, of which contain a certain level of similarity to a given B. mori protein sequence (Additional table S1, extended data (Santos, 2020)). Subsequently, we selected 1,161 gene models presenting high similarity to its respective reference protein (Identity ≥ 50%). In total, 1,094 proteins are related to the selected gene models and a small fraction of these proteins (4.6%) are the product of multiple gene models. The functional annotation pipeline was further applied using the 1,094 protein sequences, which were further assigned into different categories of their respective Gene Ontology (GO) terms using the Blast2GO software (Götz et al., 2008) (Additional table S2, extended data (Santos, 2020)). The NCBI “moth” protein database identified 1,002 proteins (91.5%) with information, and 826 (75.5%) having a gene ontology assignment. The most abundant GO categories of these proteins are shown in Figure 3.
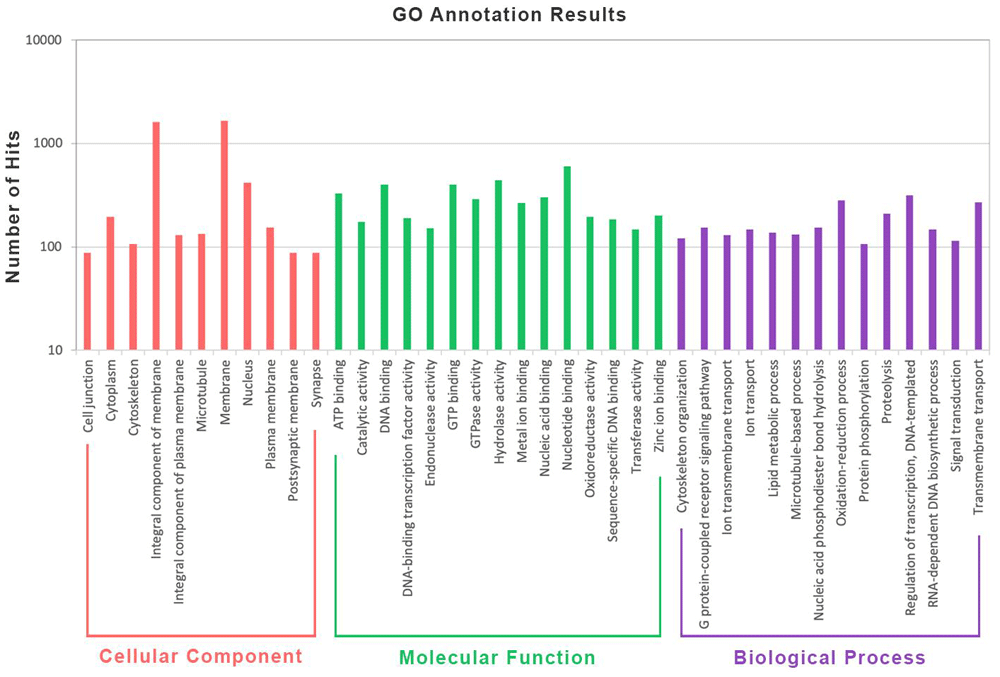
The results are categorized in three main categories: cellular component (orange), molecular function (green), and biological process (purple). The y-axis indicates the number of hits identified for each category.
The rise of low cost third generation sequencing technologies has been motivating the development of innovative genomic studies involving non-model species (Ellegren, 2014). Consequently, we notice an emergent increase in the demand for genome assemblies of more organisms. Although initial “draft” assemblies might not be contiguous enough to elucidate more complex events such as whole genome duplications, these projects have been demonstrated to be essential in the investigation of important genomic characteristics of poorly studied organisms, such as repetitive regions, genome size estimation, occurrence of variation, synteny with other species, gene size evolution and protein sequence evolution (da Fonseca et al., 2016; Fournier et al., 2017; Liyanage et al., 2019).
The key objective of this study was to provide the first genome representation of the sugarcane borer, D. saccharalis, using a high throughput sequencing approach to produce de novo genomic sequences. Our analyses indicate that the species has an estimated genome size of 359 Mb and produced a total length of 453 Mb of assembled genomic sequences representing the D. saccharalis genome. The genomic GC content was 33.74%, which is in the range of other lepidopteran species such as Ostrinia scapulalis (37.4%) (Gschloessl et al., 2018a), S. frugiperda (32.9%) (Kakumani et al., 2014), and B. mori (38.8%) (Kawamoto et al., 2019).
Repetitive element estimates from the de novo assembled sequences revealed that the D. saccharalis genome is over 40% composed of repetitive sequences. This observation, in addition to the discrepancy between the estimated genome size and the total assembly length, suggests that the assembly of repetitive-rich regions is more difficult when using short-reads, leading to the formation of an accurate but more fragmented assembly with fairly small contigs (Peona et al., 2018; Treangen & Salzberg, 2012).
The consistency of our pipeline was confirmed by analyzing the repetitiveness present in B. mori chromosomes. The percentage of repetitive elements found in this species (46.32%) was significantly similar to the findings from Kawamoto et al., 2019 (46.45%). Additionally, the high proportion of repetitiveness also appears in other lepidopteran members such as H. vespertilio (50.3%) (Pippel et al., 2020), T. pityocampa (45.3%) (Gschloessl et al., 2018b) and S. frugiperda (29%) (Gouin et al., 2017).
In terms of completeness of genic regions, our D. saccharalis assembly showed the presence of a significant number of conserved insect single-copy genes. Out of 1,658 Insecta BUSCO gene orthologs, only 3.9% were missing in the assembly. When compared to a well-studied species, B. mori, which has its genome assembled at a chromosome level, the number of missing genes is 1%, suggesting that some of these genes are likely absent from the genomes of some lepidopteran species.
Genetic similarities between the two species were also evidenced through functional annotation of predicted genes using a homology-based approach. The analyses identified over 1,000 gene models on D. saccharalis genome using protein information from B. mori. Further gene ontology annotation revealed that proteins with high identity levels between the two species are related to important cellular components such as the nucleus, plasma membranes and cytoplasm, as well as being involved in molecular mechanisms essential for cell maintenance such as the transcription process, protein synthesis and metabolic pathways. However, future additional transcriptome evidence would improve the annotation and validate these gene models, providing more solid information about the genes present in the D. saccharalis genome.
To this end, the combination of a de novo assembly algorithm and several predictive models has successfully identified important features of the D. saccharalis genome for the first time, and generated sequence data that can be immediately used at several levels: the discovery of additional genomic features, the identification of SNP markers (Boutet et al., 2016) and improvements on the genome annotation by the combination with transcriptomic data (Holt & Yandell, 2011). Furthermore, this will provide valuable resource for the investigation of potential novel techniques to control this pest on crops (Kirk et al., 2013).
Whole-genome sequencing project of Diatraea saccharalis. BioProject, Accession number: PRJNA647758. https://www.ebi.ac.uk/ena/browser/view/PRJNA647758.
Raw gDNA Illumina reads of Diatraea saccharalis. Biosample, Accession number: SAMN15594284. https://identifiers.org/biosample:SAMN15594284
The complete draft genome assembly of Diatraea saccharalis. GenBank, Accession number: JACGTY000000000. https://www.ncbi.nlm.nih.gov/nuccore/JACGTY000000000.
Zenodo: bslucas98/Dsaccharalis_genomeassembly: A first draft genome of the sugarcane borer, Diatraea saccharalis https://doi.org/10.5281/zenodo.4067084 (Santos, 2020).
This project contains the following extended data:
• Select_subset_reads.py (script used to select a subset of raw reads)
• SupplementaryTables_DsacGenome.xlsx (Additional Table 1 and 2)
• protein_identity.py (Script used to select protein gene models having >=50% similarity to the B. mori model)
This data is licensed under the terms of the Creative Commons Attribution 0 1.0 Universal (CC0).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)