Keywords
S protein, S1 subunit, betacoronaviruses, bovine coronavirus, MERS-CoV, SARS-CoV, SARS-CoV-2, phylogenetic analysis
This article is included in the Emerging Diseases and Outbreaks gateway.
This article is included in the Coronavirus (COVID-19) collection.
S protein, S1 subunit, betacoronaviruses, bovine coronavirus, MERS-CoV, SARS-CoV, SARS-CoV-2, phylogenetic analysis
According to the International Committee on Taxonomy of Viruses (ICTV; 2019, Release #35), betacoronaviruses (BetaCoVs) have been classified as belonging to Riboviria realm, Orthornavirae kingdom, Pisuviricota phylum, Pisoniviricetes class, Nidovirales order, Cornidovirineae suborder, Coronaviridae family, Orthocoronavirinae subfamily, and Betacoronavirus genus; species/subspecies of BetaCoVs are listed in Figure 1.
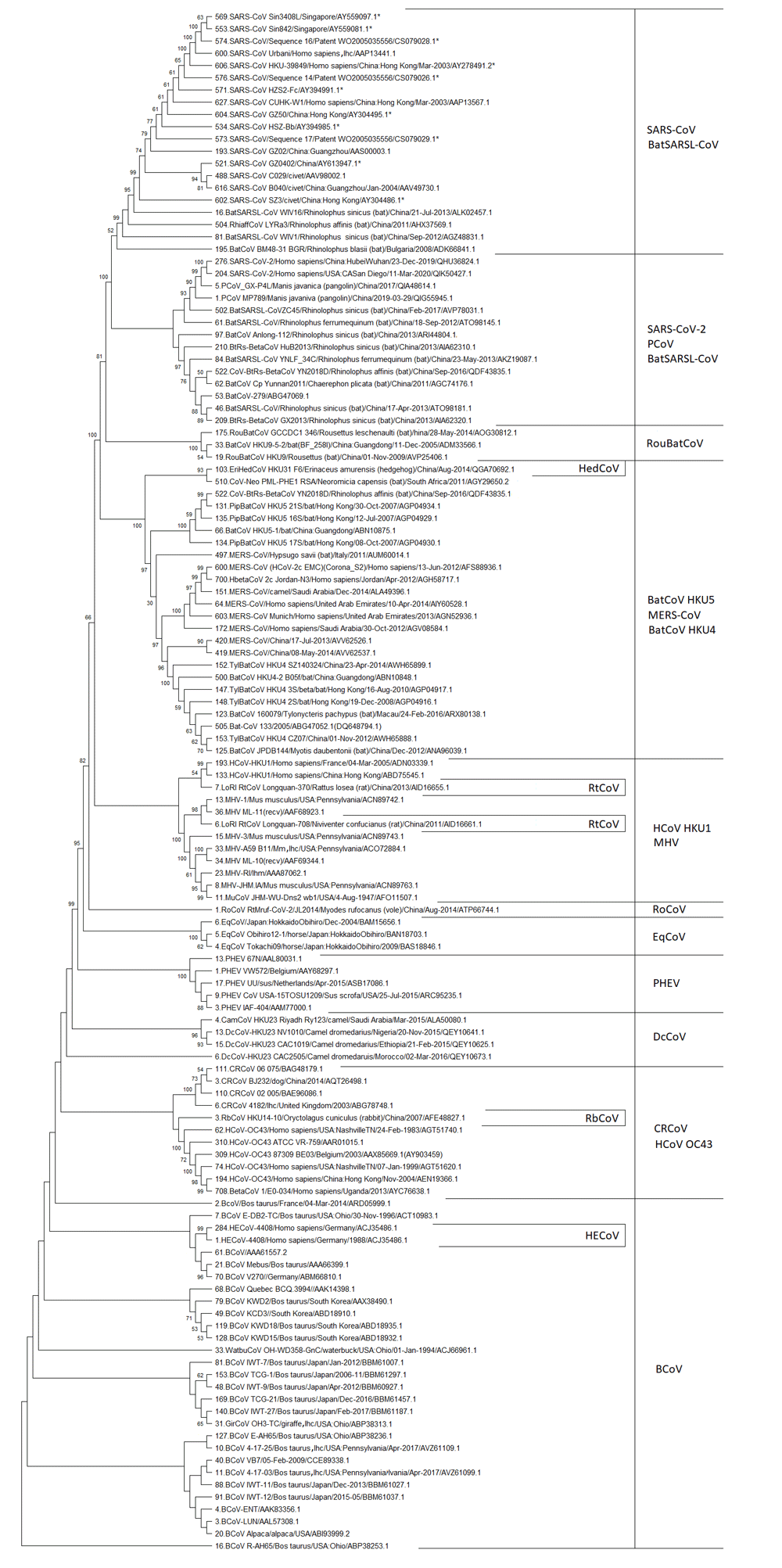
The following abbreviations are applied: lhc – the lab host cells, lhm – the lab host mouse; and SARS-CoV – Severe acute respiratory syndrome coronavirus, PCoV – Pangolin coronavirus, BatSARSL-CoV – Bat SARS-like coronavirus, BatCoV – Bat coronavirus, BetaCoV – Betacoronavirus, RoCoV – Rodent coronavirus, RtCoV – Rat coronavirus, RouBatCoV – Rousettus bat coronavirus, HCoV – Human coronavirus, HBetaCoV – Human betacoronavirus, HECoV – Human enteritis coronavirus, EriHedCoV – Erinaceus Hedgehog coronavirus, PipBatCoV – Pipistrellus bat coronavirus, HypBatCoV – Hypsugo bat coronavirus, MERS-CoV – Middle East respiratory syndrome coronavirus, TylBatCoV – Tylonycteris bat coronavirus, PhiaffCoV – Rhinolophus affinis coronavirus, CoV-Neo – Coronavirus Neoromocia, EqCoV – Equine coronavirus, MHV – Murine hepatitis virus, MuCoV – Murine coronavirus, PHEV – Porcine hemagglutinating encephalomyelitis virus, RbCoV – Rabbit coronavirus, DcCoV – Dromedary camel coronavirus, CamCoV – Camel coronavirus, CRCoV – canine respiratory coronavirus, BCoV – Bovine coronavirus, WatbuCoV – Waterbuck coronavirus, GirCoV – Giraffe coronavirus. The stars designate protein sequences deduced from nucleotide sequences using the GeneRunner program. The numbers in front of sequence annotation are the unique sequence numbers for each S/S1 sequence in the batch for each BetaCoV species for more comfortable use. Using data from 12–17.
The ongoing pandemic outbreak of coronavirus disease 2019 (COVID-19) with pneumonia symptoms has been caused by a new BetaCoV, severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2; originally named as 2019-nCoV)1,2. Two other BetaCoVs, SARS-CoV and MERS-CoV, have caused epidemic outbreaks of infectious diseases - Severe Acute Respiratory Syndrome (SARS) 2002–2003 in China and Middle East Respiratory Syndrome (MERS) in 2012 in the Middle East, respectively. All these outbreaks are severe or even fatal human diseases2,3. The other three human BetaCoVs (human coronavirus OC43, NL63, and HKU1; HCoV-OC43, HCoV-NL63, and HCoV-HKU1), usually cause cold symptoms2,3.
The rest of BetaCoVs primarily infect nonhuman mammals, among which bovine coronavirus (BCoV) is the most significant for the farming industry all over the world. Although, BCoV infected cattle have low mortality, they suffer from calf diarrhea (winter dysentery), respiratory symptoms, and substantial losses in milk and meat production4. Another BetaCoV, porcine hemagglutinating encephalomyelitis virus (PHEV), which is the causative agent of neurological and digestive disease in pigs, is also significant for farmers. Although it remains poorly studied because of its low clinical prevalence reported so far, it could lead to an animals' fatal end, causing significant harm to the swine industry5. Another BetaCoV is equine coronavirus (EqCoV), which causes diarrhea in foals and impacts the horse breeding industry6. There is also canine respiratory coronavirus (CRCoV), which is found in pet animals, associated with mild to severe canine infectious respiratory disease7.
Several small mammal BetaCoVs have been discovered to date. Among them are rodent coronavirus (RoCoV), rabbit coronavirus (RbCoV), and hedgehog coronavirus (HedCoV)8. There is also the more studied murine hepatitis virus (MHV), which causes hepatitis, enteritis, respiratory diseases, and encephalomyelitis in the central nervous system in mice and rats9. Furthermore, there are different bat coronaviruses (BatCoV), which have well-adapted hosts (different species of bats) in the natural environment. These BetaCoVs should be specially noted as they are suggested to be the origin of MERS-CoV, SARS-CoV, and SARS-CoV-210,11.
The S (spike) protein is one of three viral envelope proteins. It is considered a member of the class I viral membrane fusion proteins, including those from the influenza virus, human immunodeficiency virus (HIV), and Ebola virus. The S protein is involved in the initiation of the infectious process. It acts as an intermediary of viral and host cell membrane fusion and is a significant inducer of host immune responses12–15. The S protein assembles into trimmers and folds so that it sticks out from the membrane surface to form spikes; hence its name: spike protein. The virion surface looks like a corona (Latin for crown) because of these spikes, and this feature became the reason for the name coronaviruses12–15.
In most species of CoVs, the S protein is cleaved into two approximately equal size subunits, S1 and S2. The S1 subunit contains a host cell receptor binding domain (RBD). There are detected N-terminal domain (NTD) and C-terminal domain (CTD) in the S1 subunit, and one or both of which function as RBD. NTDs are responsible for binding the sugar (O-ac-Sia – 9-O-acetyl sialic acid) or the protein receptor CEACAM1 (the carcinoembryonic antigen-related adhesion molecule 1), and CTDs are responsible for recognizing protein receptors ACE2 (angiotensin-converting enzyme 2 – the zinc peptidase) and DPP4 (the dipeptidyl peptidase 4 – the serine peptidase), CD209L, and CD209 – the immunoglobulin-like cell adhesion molecule12–17. The schematic structure of the Betacoronavirus S protein is shown in Extended data18.
The S1 subunit is the most divergent region of the S protein, and the S1 RBD is the principal determinant of species and tissue susceptible to infection12. Therefore, its phylogenetic analysis is very important for studying coronavirus evolution. Although many phylogenetic analyzes have been reported for S or S1 on a genetic or protein level, no study had been made for all publicly available S1 protein sequences of all known BetaCoVs. In this study, the data of BetaCoV S1 protein sequence phylogenetic analysis has been presented. The S protein sequences used have been collected from GenBank before April 2020.
A total of 1595 S protein sequences, which are publicly available in GenBank from the beginning of the pandemic event to April 2020, have been used in this study. Some S protein sequences have been deduced from the corresponded nucleotide sequences using the GeneRunner program. Only 679 different of them have been implemented to the phylogenetic analysis since identical sequences do not contribute to phylogenetic relationships.
Identical S and S1 protein sequences have been found using the ClustalW option of the MEGA X (Version 10.0.5) program19 and excluded from the phylogenetic analysis. All identical S and S1 protein sequences are available as Underlying data20. S1 ends for SARS-CoV-2 (-RRAR685), SARS-CoV (-SLLR667), MERS-CoV (-RSVR751), HCoV-OC43 (-RRSR757), HCoV-HKU1 (-RRKRR760), MHV-A59 (-RRAHR721), BCoV-Quebec (-RRSRR768), BatCoV-HKU4 (-STFR749), and BatCoV-HKU-5 (-RVRR745) have been determined according to Millet and Whittaker (2014) and James et al. (2020)21,22. The rest of the S1 subunit ends have been deduced using the S1 sequence alignment in ClustalW (see Underlying data23).
Phylogenetic analysis has been performed with the MEGA X (Version 10.0.5) program19, using the Maximum Likelihood method and the JTT matrix-based model24 with 1000 bootstrap replications and uniform rates among sites. The analysis of 679 different sequences of the S1 subunit has been implemented in two steps. In the first step, different phylogenetic trees have been constructed for each BetaCoV species/subspecies, or several BetaCoV species/subspecies have been combined into the one primary tree25–39; except for human enteritis coronavirus (HECoV), for which all different protein sequences have been used in the summarised tree. RbCoV, RoCoV, rat (Rt) CoV, and HedCoV have been included together in the primary tree34. Also, SARS-CoV-2 and pangolin coronavirus (PCoV) have been combined into the one primary tree39. Bat CoVs have been divided into four groups by alignment; the primary tree has been constructed for each of them35–38. In the second step, the sequences have been selected from primary trees, and a summarized tree has been constructed. As can be seen from each phylogenetic tree (see Underlying data25–39) one or more sequences have been selected from each phylogenetic clade. The sequences have been chosen as follows: if the separated clade consists of several sequences, then the sequences that have been found closer to the branching point of the entire clade are selected; if the clade consists only of one sequence, this sequence is taken into the summary tree. The summary bootstrap consensus tree have been inferred from 1000 replicates. The percentage of replicate trees in which the associated taxa clustered together is shown next to the branches. Percentages ≥50% are shown (Figure 1).
Figure 1 shows that all S1 subunits of BetaCoVs species are originated from BCoV S1. The BCoV group also includes other ruminant BetaCoVs, which do not have an individual detached clade. There is an intermediate branch of human enteritis coronavirus among the BCoV group. It confirms the transmission of HECoV from bovine.
Furthermore, the phylogenetic clade of CRCoV and HCoV-OC43 has been separated from BCoVs, and then four clades have been detached one after another. These are Dromedary camel (Dc) CoV, PHEV, EqCoV, and RoCoV. They are followed by the clade consisting of MHV and HCoV-HKU1, which has several intermediates of RtCoVs.
After that, the group of MERS-CoV, BatCoV-HKU-4, and BatCoV-HRU-5 separate, and has an intermediate group of HedCoV. The MERS-CoV is already one of the particularly dangerous BetaCoVs for humans40. This group is followed by the group of RouBatCoVs (Rousettus bat coronaviruses).
Finally, the SARS-CoV clade is formed. It is divided into two phylogenetic branches. One consists of SARS-CoVs and BatSARSL-CoVs (Bat SARS-like coronaviruses). Another consists of SARS-CoV-2s, PCoVs, and BatSARSL-CoVs. This clade could be named as that of the pandemic BetaCoVs, because of SARS-CoV-2.
Thus, we see in Figure 1 that the evolution of BetaCoV S1 proceeds from BCoV, which is not dangerous for humans, to SARS-CoV and SARS-CoV-2, which are especially hazardous for humans.
The phylogenetic analysis carried out in this study has shown that the evolution of S1 of BetaCoVs begins from BCoV, which is not dangerous for humans, and then, passing through BetaCoVs of dogs (CRCoV), camels (DcCoV), pigs (PHEV), horses (EqCoV), rodents (RoCoV, MHV, RtCoV) and hedgehogs (HedCoV) leads to SARS-CoV and SARS-CoV-2, which are already particularly dangerous for humans. Therefore, we shouldn't underestimate the potential danger of BCoV.
Figshare: 100% homology sequences of Betacoronaviruses S protein and S1 subunit, https://doi.org/10.6084/m9.figshare.12962378.v420.
Figshare: The S1 sequence alignment of Betacoronaviruses, https://doi.org/10.6084/m9.figshare.1310689423.
Figshare: The phylogenetic analysis of the Bovine coronavirus S1 subunit protein sequence, https://doi.org/10.6084/m9.figshare.12956963.v525.
Figshare: The phylogenetic analysis of the Human coronavirus S1 subunit protein sequence, https://doi.org/10.6084/m9.figshare.12957932.v426.
Figshare: The phylogenetic analysis of the Severe Acute Respiratory Syndrome-related coronavirus S1 subunit protein sequence, https://doi.org/10.6084/m9.figshare.12957977.v527.
Figshare: The phylogenetic analysis of the Middle East Respiratory Syndrome coronavirus S1 subunit protein sequence, https://doi.org/10.6084/m9.figshare.12957989.v428.
Figshare: The phylogenetic analysis of the Murine hepatitis virus S1 subunit protein sequence, https://doi.org/10.6084/m9.figshare.12958004.v429.
Figshare: The phylogenetic analysis of the Canine respiratory coronavirus S1 subunit protein sequence, https://doi.org/10.6084/m9.figshare.12958028.v430.
Figshare: The phylogenetic analysis of the Porcine hemagglutinating encephalomyelitis virus S1 subunit protein sequence, https://doi.org/10.6084/m9.figshare.12958091.v431.
Figshare: The phylogenetic analysis of the Equine coronavirus S1 subunit protein sequence, https://doi.org/10.6084/m9.figshare.12958112.v432.
Figshare: The phylogenetic analysis of the Dromedary camel coronavirus S1 subunit protein sequence, https://doi.org/10.6084/m9.figshare.12958136.v433.
Figshare: The phylogenetic analysis of the small mammal BetaCoV coronavirus S1 subunit protein sequence, https://doi.org/10.6084/m9.figshare.12958172.v434.
Figshare: The phylogenetic analysis of the Bat coronavirus (HKU3 group) S1 subunit protein sequence, https://doi.org/10.6084/m9.figshare.12958184.v435.
Figshare: The phylogenetic analysis of the Bat coronavirus (HKU4 group) S1 subunit protein sequence, https://doi.org/10.6084/m9.figshare.12958193.v536.
Figshare: The phylogenetic analysis of the Bat coronavirus (HKU5 group) S1 subunit protein sequence, https://doi.org/10.6084/m9.figshare.12958196.v537.
Figshare: The phylogenetic analysis of the Bat coronavirus (HKU9,10 group) S1 subunit protein sequence, https://doi.org/10.6084/m9.figshare.12958208.v438.
Figshare: The phylogenetic analysis of the Severe Acute Respiratory Syndrome-related coronavirus 2 S1 subunit protein sequence, https://doi.org/10.6084/m9.figshare.13102889.v239.
Figshare: The schematic structure of the betacoronavirus S protein, https://doi.org/10.6084/m9.figshare.12951413.v218.
Data are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)