Keywords
single cell, scATAC-seq, pseudoalignment
single cell, scATAC-seq, pseudoalignment
Recent technological advances in single-cell technologies resulted in a tremendous increase in the throughput in a relatively short span of time1. The increasing number of cells that could be analyzed prompted a better usage of computational resources; this has been especially true for the post-alignment and quantification phases. As a consequence, it is today feasible to run the analysis of single cell data on commodity hardware with limited resources2, even when the number of observables is in the order of hundreds of thousands. Conversely, the analysis steps from raw sequences to count matrices lagged for some time; alignment to the reference genome or transcriptome is largely dependent on classic aligners, without any specific option to handle single-cell data, with the notable exception of the latest implementation of STARsolo in the STAR aligner3.
More recently, analysis of NGS data benefit from technologies based on k-mer processing, allowing alignment-free sequence comparison4. Most of these technologies require a catalog of k-mers expected to be in the dataset and, hence, subject of quantification. RNA-seq analysis relies on the quantification of gene/transcript abundances and, while it is possible to perform de novo characterization of unknown species in every experiment, it is common practice5,6 to rely on a well-defined gene model such as GENCODE7 to quantify expressed species. It is then possible to efficiently perform alignment-free analysis on transcripts to quantify gene abundances and, in fact, tools implementing this approach such as kallisto8 or Salmon9 have been quickly adopted on a wide scale. Moreover, a recent implementation of kallisto extended its capabilities to the analysis of single cell RNA-seq data10 by direct handling of cell barcodes and UMIs, allowing the analysis of such data in a streamlined way.
Analysis of epigenetic features by ATAC-seq requires the identification of enriched peaks along the genome sequence. This is typically achieved using peak callers such as MACS11, opportunely tuned. Since ATAC-seq signal mirrors DNA accessibility as mapped by DNase-seq assays12 and catalogs of DNase I Hypersensitive Sites (DHS) are available13,14 it should be possible to perform reference-based ATAC-seq analysis in a way much similar to what is performed for RNA-seq analysis. In this paper we show it is indeed possible to perform single-cell ATAC-seq analysis using kallisto and bustools, with minor tweaks, using an indexed reference of ~1 million known DHS sites on the human genome.
Single cell ATAC-seq data were downloaded from the 10x Genomics public datasets (https://support.10xgenomics.com/single-cell-atac/datasets/1.1.0/atac_v1_pbmc_10k) and include sequences for 10k PBMC from a healthy donor. We used the Peak by cell matrix HDF5 (filtered) object as our ground truth.
We downloaded the DNase I Hypersensitive Sites (DHS) interval list for hg19 genome from the Regulatory Elements DB15, intervals closer than 500bp were clustered using bedtools16.
We extracted DNA sequences for DHS intervals and indexed corresponding fasta files using kallisto index (v0.46.0) with default parameters, resulting in an index for the full DHS set (iDHSfull) and an index for the merged set (iDHS500). The same procedure was performed for the peak set identified by cellranger-atac and distributed along with the data (iMACS).
kallisto requires the definition of the unique molecular identifiers (UMI) and cellular barcodes (CB) in a specific fastq file. For standard Chromium scRNA-seq data, these are substrings of R1 and RNA is sequenced in R2. Chromium scATAC-seq reads are not structured in the same way, paired end genomic reads are in R1 and R3, R2 includes only the 16bp cellular barcode. In addition, kallisto bus expects only a single read with genomic information. Therefore we simulated appropriate structures in three different ways:
1. by adding 12 random nucleotides and mapping the R1 file (forward read):
kallisto bus -x 10xV2 modified_R1.fastq.gz
pbmc_10k_R1.fastq.gz
2. by extracting sequences of different length n (5, 10, 15, 20) from the 5' of R3 (reverse read) and mapping the R1 file:
kallisto bus -x 1,0,16:2,0,n:0,0,0
pbmc_10k_R1.fastq.gz
pbmc_10k_R2.fastq.gz
pbmc_10k_R3.fastq.gz
3. by extracting sequences of different length n (5, 10, 15, 20) from the 5' of R1 and then mapping the R3 file:
kallisto bus -x 1,0,16:2,0,n:0,0,0
pbmc_10k_R3.fastq.gz
pbmc_10k_R2.fastq.gz
pbmc_10k_R1.fastq.gz
We will refer to the second set of simulation as n-fwd and to the third set as n-rev, where n is the number of nucleotides considered as UMI. We also applied two different summarization strategies for bustools count step. In the first approach, pseudocounts are not summarized, the number of features matches the size of the index; in the second approach, summarized, we let bustools map counts on iDHSfull to the merged intervals (Figure 1A).
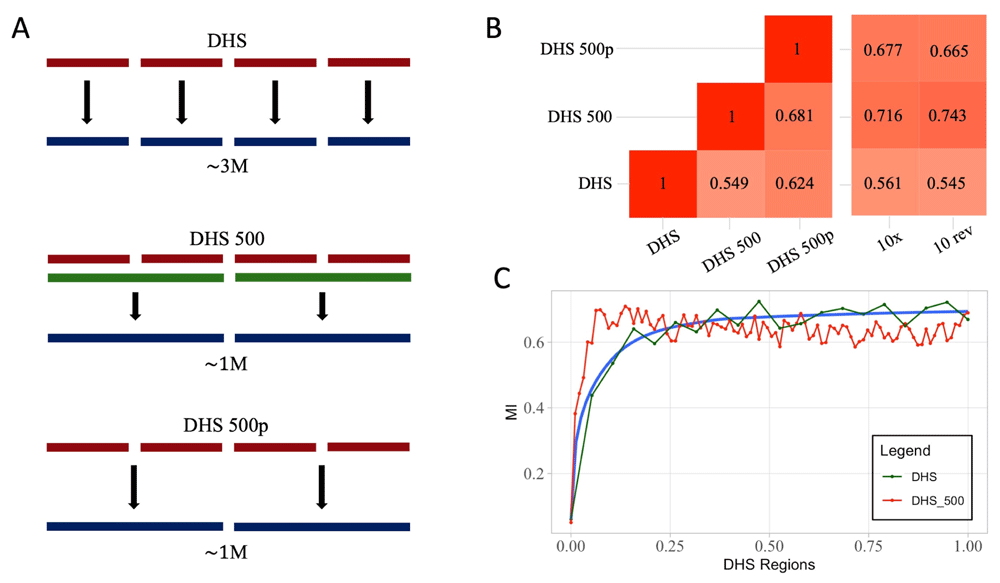
(A) Graphical depiction of processing of pseudoalignment over DHS, based on three DHS derived indices. The first (DHS) generated by kallisto on ~2M DNase I sites, the second (DHS500) by merging regions closer than 500bp and the last (DHS500p) by projecting the result of DHS index to DHS500 using bustools capabilities. (B) Heatmaps representing MI scores for the DHS derived matrices. The heatmap on the left reports the pairwise MI values between DHS, DHS500 and DHS500p strategies. The heatmap on the right represents MI values comparing the DHS derived strategies to the cellranger-atac (10x) results or 10-rev strategy. DHS500 strategy achieves the highest scores. (C) MI values comparing DHS (green line) and DHS500 (red line) strategies to cellranger-atac at different thresholds on the number of regions considered in the analysis. When approximately 50,000 regions are included, the MI stabilizes at its maximum.
Counts matrices were analysed using Scanpy (v1.4.2)2 with standard parameters. We filtered out cells that had less than 200 regions and regions that were not at least in 10 cells. The count matrices were normalized and log transformed. The highly variable regions were selected and the subsetted matrices processed to finally clusterized the data with the Leiden algorithm17. Adjusted mutual information (MI) was used to evaluate the concordance between the 10x and our matrices.
The matrices derived from kallisto and cellranger-atac were also imported into Seurat V318. Gene activity score was calculated using the CreateGeneActivityMatrix function or directly summarized by kallisto. The annotated 10k PMBC scRNA-seq Seurat object was downloaded from the link available in their v3.1 ATAC-seq Integration Vignette (https://satijalab.org/seurat/v3.1/atacseq_integration_vignette.html).
Cell labels from the scRNA-seq data were transferred using TransferData function based on the gene activity score. All the analyses were carried out using standard parameters. Jaccard similarities were evaluated using the scclusteval (v0.1.1) package19.
At time of writing, kallisto does not natively support scATAC-seq analysis, though it can be applied to any scRNA-seq technology which supports CB and UMI. According to the kallisto manual, the technology needs to be specified with a tuple of indices indicating the read number, the start position and the end position of the CB, the UMI and the sequence respectively. In this sense, the technology specifier for standard 10x scRNA-seq with v2 chemistry is 0, 0, 16 : 0, 16, 26 : 1, 0, 0 (see kallisto manual for details). Using this logic, a single fastq file contains sequence information and UMI is always required. scATAC-seq from 10x genomics is typically sequenced in paired-end mode and, moreover, there is no definition of UMI as reads can be deduplicated after genome alignment.
kallisto requires an index of predefined sequences, typically transcripts, to perform pseudoalignment and, if applied to scATAC-seq analysis, does not allow for any typical analysis in the epigenomic protocols, including the identification and quantification of enriched regions. Therefore, we computed an index on the genomic sequences for the 80, 234 peaks identified by cellranger-atac and distributed together with fastq files. This ensures that the subsequent analysis were performed on the same regions and allowed us to quantify the bias, if any, introduced by kallisto.
We tested different strategies to overcome the technical limits and the absence of UMI. We evaluated concordance of different approaches in terms of adjusted mutual information (MI) of cell groups identified with a fixed set of filtering and processing parameters. Analysis based on cellranger-atac results is considered as ground truth. Results are reported in Table 1.
The table reports adjusted Mutual Information between single cell cluster assignments on cellranger-atac data and kallisto analysis. Different strategies to evaluate pseudoUMI are reported. All simulations raised high MI values, both in the forward and reverse approach, except for the pseudoUMI of length 5. The 10-Reverse configuration reached the highest score.
| Comparison | Forward | Reverse |
|---|---|---|
| 10x vs 5nt | 0.1854 | 0.1733 |
| 10x vs 10nt | 0.7434 | 0.7625 |
| 10x vs 15nt | 0.7571 | 0.7398 |
| 10x vs 20nt | 0.7356 | 0.7520 |
| 10x vs Random | 0.7272 | None |
We tested two main strategies: in the first the R1 is pseudoaligned and the initial nucleotides of R2, cut at different thresholds, are used as UMI (pseudoUMI hereafter). As UMI is needed for deduplication, we reasoned that a duplicate in scATAC-seq should be identified by the same nucleotides, especially in the first portion of the read, where quality is higher. We observe generally high values of MI, with the notable exception of pseudoUMI 5nt long. Since basecall qualities are generally higher for R1 and kallisto does not use qualities in pseudoalignment, we also tested the strategy in which R2 is used for pseudoalignment and R1 is used to obtain pseudoUMI. Also in this scenario, 5nt pseudoUMI raised the worst results, while MI values were slightly higher than the forward configuration. In particular, we noticed the highest MI values when R2 is used and pseudoUMI is 10nt long (MI = 0.7625). We also tested a configuration using R1 as sequence and 10nt UMI randomly generated. Interestingly, concordance remains in line with previous experiments (MI = 0.7272).
These data indicate that kallisto is able to properly quantify enrichments in scATAC-seq and does not introduce a considerable bias.
As one major limitation of a kallisto-based approach to scATAC-seq is the lack of peak calling routines and the need of a index of sequences for pseudoalignments, we reasoned that we could use any collection of regions that putatively would be target of ATAC-seq experiments. Since ATAC-seq is largely overlapping DHS we exploited the regions defined in the ENCODE project20. The DHS data provided by ENCODE includes 2, 888, 417 sites. We generated an additional dataset by merging regions closer than 500bp into 1, 040, 226 sites. We performed pseudoalignment on the full dataset, on the merged dataset and, lastly, on the full dataset summarized to the merged by bustools (see Methods). Pairwise comparison between performances of the three methods reveals lower values of MI (Figure 1B). Comparison with 10x data and the configuration 10-rev previously performed shows high values of MI when considering merged DHS intervals (MI = 0.7164 and 0.743 respectively). When pseudoalignmets are performed on the full DHS set, performance degrades to less than optimal levels. Since the number of DHS intervals is considerably higher than the typical number of regions identifiable by ATAC-seq, we tested the trend of MI at different cutoffs on the number of DHS included in the analysis (Figure 1C). MI reaches a plateau when approximately 50, 000 regions are included into the analysis. This sets a reasonable target for region filtering during preprocessing stages of scATAC-seq data. In all, these findings support the suitability of using kallisto for identification of cell identities in scATAC-seq without any prior knowledge of the epigenetic status of single cells.
A crucial step in the analysis of scATAC-seq data is the identification of marker peaks which can be used to functionally characterize different clusters. We tested the ability of our reference-based approach to identify differential DNase I hypersensitive sites that are overlapping or close to peaks identified with standard analysis. To this end, we first matched cell groups from DHS500 to groups identified after cellranger-atac. We selected the top 1, 000 peaks marking each DHS500 group and evaluated the concordance by mutual distance to the top 1, 000 significant markers in the matched groups (p < 0.05), we could identify significant markers only in five matched clusters. We found that the large majority of peaks (>= 80%) were overlapping between the two strategies or closer than 20kb (Figure 2). These results confirm the substantial equivalence between the standard strategy and the reference-based one.
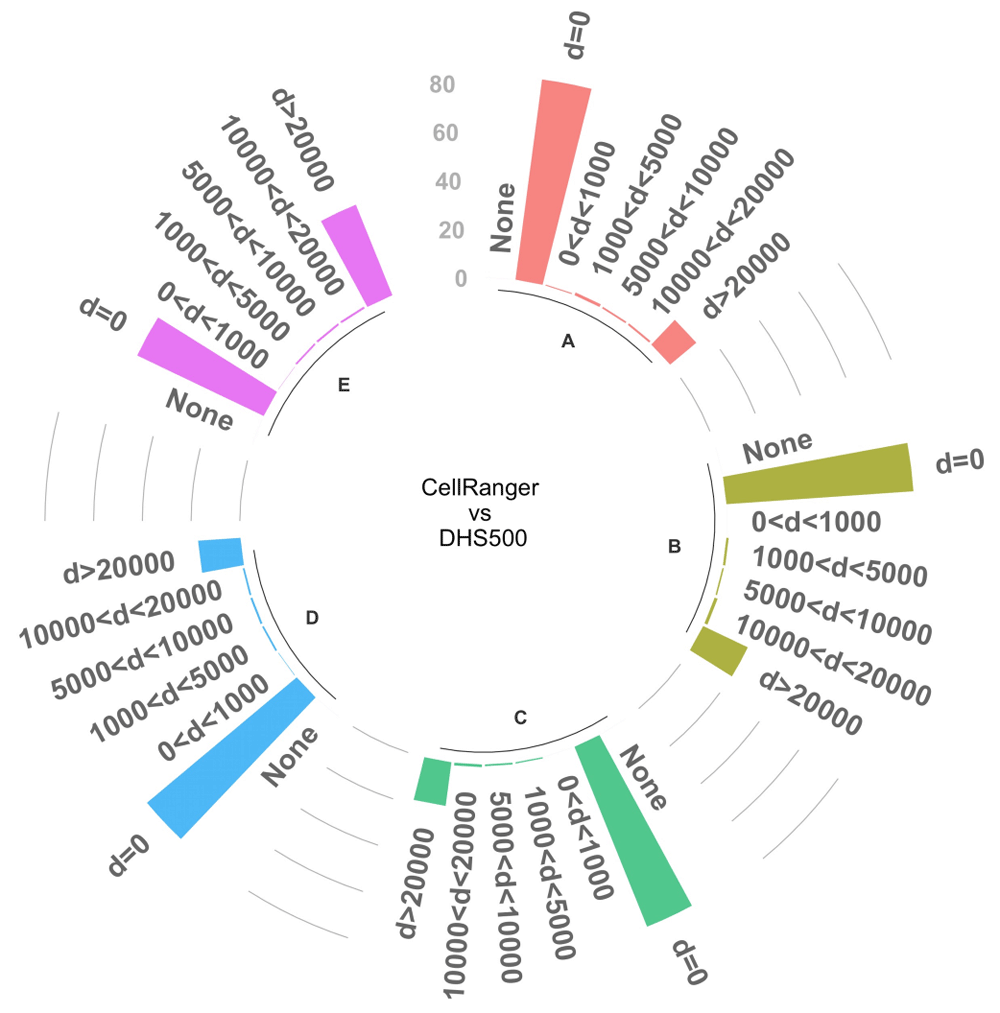
The bars represent the proportion of marker peaks that are in common between DHS500 and cellranger-atac-based strategies at different distance thresholds. Only the top 1, 000 significant peaks (p < 0.05) were included in the analysis; the graph reports results for the 5 cell clusters (A–E) that contain the required amount of significant markers. The chart also reports the proportion of peaks without any match (None).
In addition to the analysis of technical suitability of kallisto for the analysis of scATAC-seq data, we investigated its validity in extracting biological insight. To this end, we performed a more detailed analysis of PBMC data by label transferring using Seurat V318, with the hypothesis that different approaches could lead to mislabeling of cells clusters. Matching is performed with the help of Gene Activity Scores calculated as sum of scATAC-seq counts over gene bodies extended 2kb upstream the TSS, Seurat’s default approach. We applied the same transferring protocol on data derived from cellranger-atac counts and from the DHS500 approach (Figure 3), founding no relevant differences in the UMAP embeddings. A detailed quantification of cluster matches reveals a slight deviance in the characterization of NK subpopulations (Figure 4A). In addition to scores calculated by Seurat, we tested the ability of bustools summarization step to project and sum scATAC-seq values into Gene Activity using the identical mapping to extended gene bodies. In terms of cell labeling, this approach is equivalent to Seurat (Figure 4B), with the additional advantage of reduced run times.
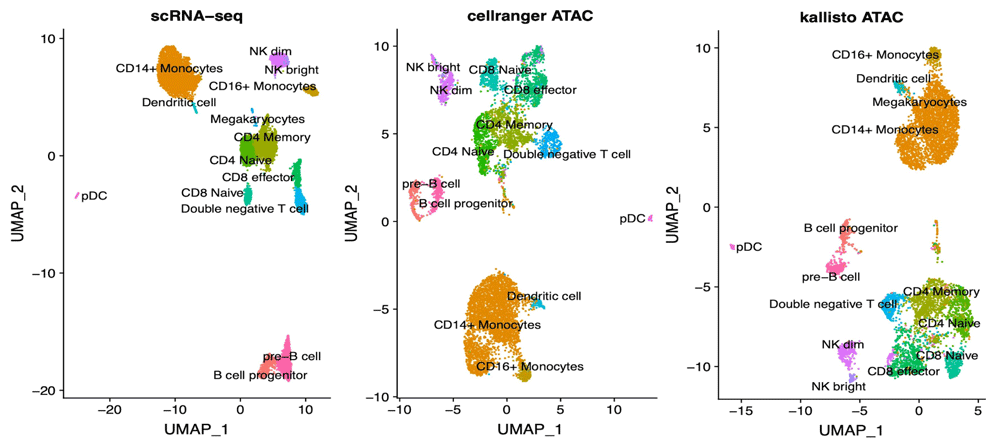
The UMAP plot on the left represents scRNA-seq data of 10k PBMC as returned by Seurat vignette. The UMAP plots in the middle and on the right represent scATAC-seq analysis on cellranger-atac or kallisto analysis respectively. Cell clusters are consistent in their topology in the three plots, indicating the validity of kallisto for this kind of analysis.
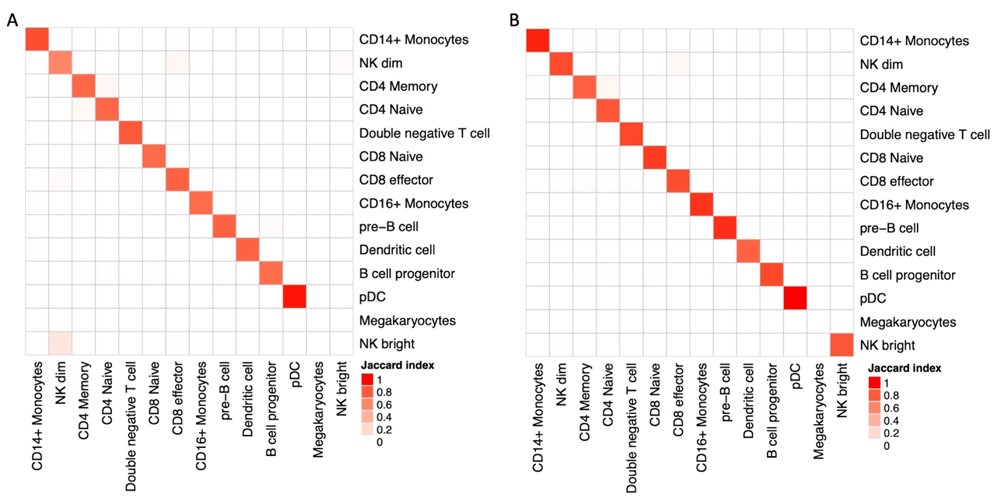
(A) Pairwise Jaccard similarity between cell annotations as a result of label transfer from RNA-seq data using Gene Activity Score evaluated by Seurat. Concordance between results after cellranger-atac (rows) and DHS500 (columns) are largely comparable, with the notable exception of NK subpopulations. (B) Pairwise Jaccard similarity between cell annotations on DHS500 when Gene Activity Score is computed by Seurat (rows) or by bustools summarization step (columns).
Analysis of differential chromatin properties, through ATAC-seq and other quantitative approaches, relies on the identification of peaks or enriched regions, this is often achieved with the same statistical framework used in analysis of differential gene expression21,22. Identification of peaks is a key difference between the two approaches, de novo discovery of unannotated transcripts has been shown to be possible in early times of NGS23, but the large majority of analysis is performed on gene models; conversely, analysis of epigenomes involves identification of regions of interest, although a large catalogues of such regions have been provided by several projects, such as the ENCODE project24, the BluePrint project25 or the GeneHancer database26. In single cell analysis, both scRNA-seq and scATAC-seq, identification of novel features may be an issue, especially because of the low coverage at which single cells are profiled. This work is the first, to our knowledge, to test the feasibility of a reference-based approach to ATAC-seq analysis, with a special focus on single cell ATAC-seq. In combination, we tested the suitability of kallisto to this kind of analysis, to maximise the performances of the whole process. Our results suggest that identification of cell groups using a reference-based approach is not different from a standard pipeline. Not only cells could be classified in a nearly identical way, but also differential features are largely matched between the analysis. The most obvious advantage is the gain in speed and efficiency: once reads have been demultiplexed, kallisto analysis requires short execution times, in the order of minutes, with limited hardware resources; this advantage has been known for a while and, in fact, it is has been demonstrated that it can be used on Rock64 hardware27. We also anticipate that adoption of a reference-based strategy comes with additional advantages, in particular functional annotations and gene associations are available for known regulatory regions20 and, more recently, for DNase I Hypersensitive Sites14. Of course, our strategy has some limitations that come from the unavailability of read positioning on the genome: in addition to the impossibility of identifying novel peaks, it is not possible to perform some ATAC-specific analysis, such as nucleosome positioning or footprinting of transcription factors in accessible regions. Indeed, these two can be overcome if standard alignment is used in place of pseudoalignment, at cost of time. As concluding remark we would like to underline that, although we showed that kallisto can be effectively used for analysis of scATAC-seq data, we are aware that it has not been conceived for that purposes and, indeed, its interface needs some tweaks to make it work; for this reason we advocate the development of tools which support scATAC-seq natively and other tools for postprocessing and data visualization.
Single cell ATAC-seq data were downloaded from the 10x Genomics public datasets (https://support.10xgenomics.com/single-cell-atac/datasets/1.1.0/atac_v1_pbmc_10k) and include sequences for 10k PBMCs from a healthy donor. Access to the data is free but requires registration.
Zenodo: vgiansanti/Kallisto-scATAC v1.0. https://doi.org/10.5281/zenodo.370317428.
This project contains a detailed explanation of the procedures described in this work and the list of DHS sites; this is also available at https://github.com/vgiansanti/Kallisto-scATAC.
Extended data are available under the terms of the Creative Commons Attribution 4.0 International license (CC- BY 4.0).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)