Keywords
High-Throughput Sequencing, Alignment, Sorting, High-Performance Computing, MPI
High-Throughput Sequencing, Alignment, Sorting, High-Performance Computing, MPI
Since the first next generation sequencing technology was released in 2005 (Kchouk et al., 2017), considerable progress has been made in terms of sequencing quality, diversity of protocols and throughput of the machines. As of today, the most recent generation of sequencers can easily produce terabytes of data each days and we expect this exponential growth of the sequencing to continue. This data tsunami raises many challenges, from data management to data analysis, requiring an efficient high-performance computing architecture (Lightbody et al., 2019). Indeed, the throughput capacity of the sequencers tends to overwhelm the capacity of common computer architectures and the data analysis workflow to handle such amount of data in a reasonable time. As we have entered the era of genomic medicine, delivering the results to the clinicians within a short delay to guide the therapeutic decision is a challenge of the utmost importance in daily clinical practice. Several national initiatives worldwide such as France, USA, UK or Australia (Stark et al., 2019) promote the use of genomics into healthcare. There is no doubt that exascale informatic architecture and software are required to tackle the challenges raised by the genomic medicine.
A typical bioinformatics workflow to analyze high-throughput sequencing (HTS) data consists of a set of systematic steps of pre-processing to i) align (or map) the sequencing reads on a reference genome and ii) to sort the alignments according to their coordinates on the genome. Those steps are fundamental for the efficiency and relevance of the downstream analysis to decipher genomic alterations such as mutations or structural variations. Traditional tools for aligning the reads are BWA-MEM (Li & Durbin, 2010) and SOAP2 (Li et al., 2009a), and the sorting is usually performed with Samtools (Li et al., 2009b), Sambamba (Tarasov et al., 2015), Picard tools and GATK (McKenna et al., 2010). Most of the time, these steps are very time consuming (up to several days for whole genome analysis) as they suffer from bottlenecks at the CPU, IO and memory levels. Therefore, removing these bottlenecks would make it possible to reduce the time-to-delivery of the results such that they could be available within a reasonable delay when very large data are produced by the sequencers.
In order to tackle the aforementioned challenges to align and sort the sequencing data, we developed software based on the Message Passing Interface (MPI) communication protocol (Gropp et al., 1996) that makes it possible to fully benefit of parallel architecture of supercomputers Hupé & Jarlier (2020a; 2020b). MPI can therefore reduce the different bottlenecks by capitalizing on the low-latency network fabrics generally available on modern supercomputers. This allows an efficient distribution of the workload over the available resources of the supercomputers thus providing the expected scalability.
For the alignment, the software consists of a MPI wrapper to the BWA-MEM software such that the original code remains unchanged. The wrapper uses parallel IO and shared memory (Figure 1). It first parses the input FASTQ files in order to define chunks of equal size. The different chunks are actually represented as two offsets (start and end) from the original FASTQ file thus reducing the amount of information to store. The reference genome is loaded into the shared memory on each computer node. Then, each chunk is processed in parallel threads by the original BWA-MEM algorithm. The results is finally written thanks to a shared file pointer in a unique SAM file. For a human genome, the size of the reference genome file in the shared memory takes around 5 GB and the total memory used by a BWA-MEM thread is around 300 MB. The original BWA-MEM processes a chunk of reads at a time that contains the same number of nucleotides which is also the case with our parallel implementation in order to allow the reproducibility with the original algorithm. We name our code alignment mpiBWA (Hupé & Jarlier, 2020a).
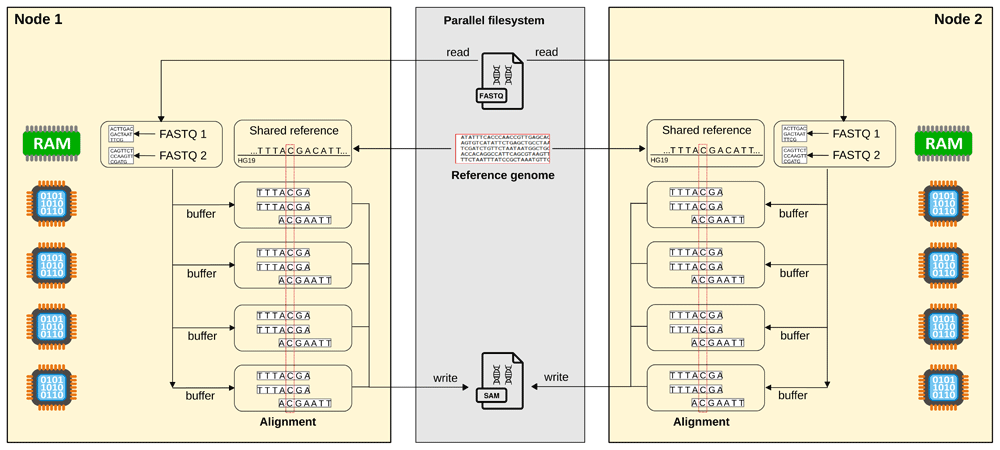
Each core is in charge of aligning a chunk of the FASTQ file. The reference genome is stored in shared memory of each node of the computing cluster. Once aligned, the results are written into a SAM file. To ensure scalability, the read and write operation require a parallel filesystem.
For the sorting, the software implements a parallel version of the bitonic sort proposed by Batcher (1968) for sorting any sequence of elements of size n = 2k being of power of 2. Its complexity is 𝒪 (n log2n) that is higher than 𝒪 (n log n) from the popular merge sort algorithm. However, the bitonic sort is very suitable for parallel implementation since it always compares elements in predefined sorting network that is independent of the input data. To understand how the algorithm works, we first defined in what follows some concepts (see Grama et al. (2003) for details).
The algorithm relies on a bitonic sequence that is a sequence of values 〈a0, a1, … , an−1〉 with the property that 1) there is an index i, 0 ≤ i ≤ n−1 such that 〈a0, a1, … , ai〉 is monotonically increasing and 〈ai+1, … , an−1〉 is monotonically decreasing, or 2) there exists a cyclic shift of indices so that the condition 1) is satisfied. From any bitonic sequence s = 〈a0, a1, … , an−1〉, the bitonic split operation consists in transforming the input sequence s into these two subsequences:
Batcher (1968) proved that both s1 and s2 are bitonic sequences and the elements of s1 are smaller than the elements of s2. Thus, a recursive bitonic split from a bitonic sequence of size n = 2k , until the sequence obtained are of size one allows the sorting of the input bitonic sequence in k splits as shown in Figure 2. The procedure of sorting a bitonic sequence from bitonic split operation is called bitonic merge. For each split of a bitonic merge, n/2 comparisons are performed during which the two numbers are exchanged if not in the right order using a compare-exchange operation. A bitonic merge procedure of a sequence of size n is noted if the comparisons sort the number in monotonically increasing order or for monotonically decreasing order.
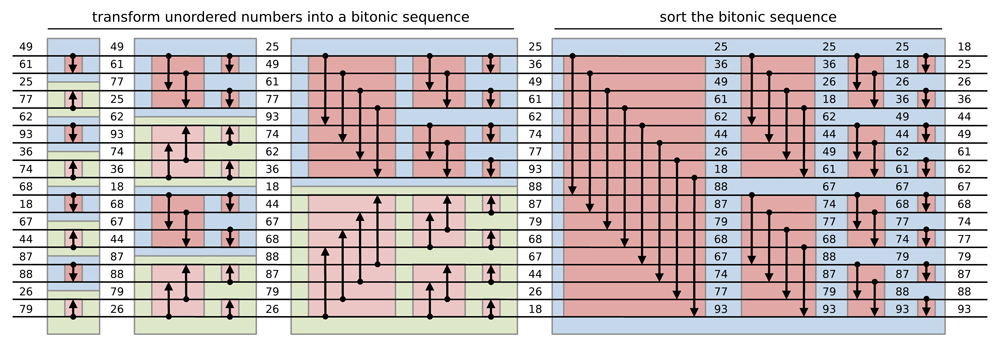
Each horizontal line represents an input and each arrow represents a two-input/two-output comparator which performs compare-exchange operation. Depending on its direction, the outputs are sorted in increasing (decreasing) order with an downwards (upwards) arrow. First, the comparator network transforms an input sequence of 16 unordered numbers into a bitonic sequence alternating bitonic merges. Then, the obtained bitonic sequence is sorted with a recursive bitonic merge procedure (adapted from Grama et al. (2003)).
Sorting any sequence of unordered number thus requires to convert the input sequence into a bitonic sequence. This is obtained using a bitonic sorting network (see Figure 2) that sorts n = 2k numbers using k − 1 bitonic sorting stages, where the i-th stage is composed of n/2i alternating increasing and decreasing bitonic merges. The final stage being to obtain the sorted sequence.
It is straightforward to generalize the bitonic sort algorithm on parallel architecture as shown in Figure 3. Each processor is in charge of an even number m = n/p elements (where p is the number of processors that is a power of 2) that are first sorted using a merge sort algorithm. Then, each comparison over the bitonic sorting network is replaced by a pair of processors with performs a compare-split operation. During the compare-split operation, the two sorted sequences from each processor are merged into one monotonic sorted list, then bisected into two (lower and higher) sequences. After the compare-split, one processor will keep the lower m elements from sequence and the other processor will keep the higher m elements according to the direction of the arrow in the step (see Kim et al. (2001) & Grama et al. (2003) for details). This parallelization allows the sorting of n elements in 𝒪 (log2n) time using a bitonic sorting network.
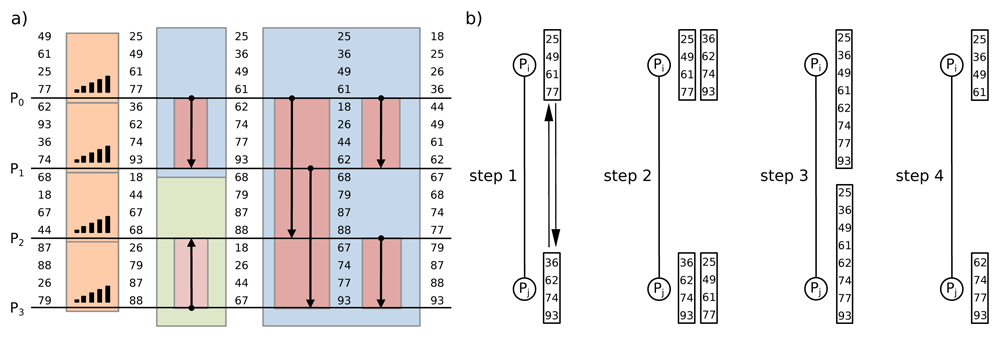
(a) Each horizontal line represents an input of elements attached to a processor. Each processor first locally sorts its list of 4 elements, then, the parallel sort performs three steps of compare-split operations over the bitonic sorting network. (b) During the compare-split operation, each pair of processors first exchanges their information: each processor sends its block to the other process, then it merges the two sorted blocks into one list and stores only the appropriate half of the merged block to be used in the next step (adapted from Kim et al. (2001) & Grama et al. (2003)).
The workflow for the sorting is described in Figure 4. The SAM file is read and split into p blocks that are dispatched across the p processors. Each block is parsed such that for each line of the SAM file, we extract the genomic coordinates of each read, its sequence (and all other information contained in the SAM file) and the line is indexed according to its offset in the SAM memory buffer of a processor. Then, the genomic coordinates are sorted with the bitonic sort as shown in Figure 3. During the sorting, a vector with five values follows the bitonic sorting network including the genomic coordinate c, the rank ri of the processor that parsed the block of the input SAM file, the offset oi of the line from the input SAM file, the rank rd of the processor that will write the block of the sorted data in the destination SAM file and the offset od of the line in the sorted destination SAM file. Obviously, the values rd and od are known when the bitonic sort is completed. It is important to highlight that the parallel computation is performed by different processors that are located on different compute nodes. This means that a block of data that has been read by a given processor is not accessible by another processor. Moreover, to optimize the writing of the sorted destination SAM file, it is essential to write the data in contiguous block. Thus, it is necessary that a processor in charge of the writing owns locally the data from a block of contiguous offsets od in the sorted destination file. This implies that all the data have to be shuffled across all the processors that have to exchange data in an all-to-all communication step. To optimize the communication between the processors during the shuffle, we have implemented a Bruck algorithm (Bruck et al., 1997) of time complexity 𝒪 (log2 p). The Bruck algorithm is performed twice as shown in Figure 4 and can be seen as a joint procedure between two tables (the elements of the table being located on different processors). During the first Bruck phase, the rd and od are sent back to the corresponding processor from where the data originated, and then, during the second Bruck phase, all the data (i.e. the appropriate lines of the SAM file) are sent to the processor that has been assigned to write the data. During the second phase the original data is copied into the memory buffer and then exchanged.
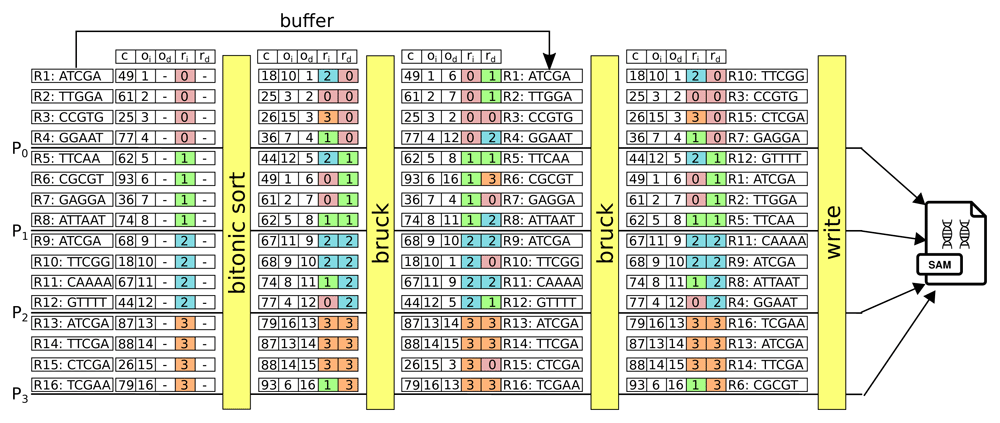
The SAM file is read and split over the 4 processors P0 to P3 that sort the data with the bitonic sort. Then, a first Bruck phase sent the rd and od values back to processor from where the data originated. The data from the SAM file are sent by a second Bruck phase to the processor that has been assigned to write the contiguous blocks (during this step, the original data is copied into the memory buffer and then exchanged). Finally, the data can be written on a parallel filesystem. c: genomic coordinate. ri: rank of the processor that parsed the block of the input SAM file. oi: offset of the line from the input SAM file. rd: rank of the processor that will write the block of the sorted data in the destination SAM file. od: offset of the line in the sorted destination SAM file.
Note that when the SAM file contains several chromosomes, each chromosome is sorted successively. Therefore, the upper memory bound depends on the size of the whole SAM file plus internal structures plus the size of the biggest chromosome. In this case, the total amount of memory used is around 1.5 the size of the original SAM file if the file contains read on all the chromosome of the human genome. If the SAM file contains only one chromosome, then the total amount of memory required is 2.5 the size of the original SAM file. To be efficient, the sorting requires a number of cores being a power of 2. We name our code for sorting mpiSORT (Hupé & Jarlier, 2020b).
We benchmarked mpiBWA and mpiSORT on the HG001 NA12878 sample from GIAB (Zook et al., 2014). This sample is a whole genome with 300X depth of coverage composed of 2.13 billions 2x250 pair-ended reads. The BAM file has been downloaded from:
url: ftp://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/NA12878/
folder: NIST_NA12878_HG001_HiSeq_300x/NHGRI_Illumina300X_novoalign_bams/
The BAM file has been converted into FASTQ files using bedtools bamtofastq. In order to test the scalability of our software with respect to the size of the data, we downsampled the original 300X sample to obtain FASTQ files corresponding to depths of coverage ranging from 28X to 300X with a geometric progression with a common ratio of 1.6. The sequences have been aligned on the human genome GRCh38.
We ran the benchmark on a computing cluster equipped with Intel Xeon® Gold 6148 CPU @ 2.40GHz (Skylake architecture). The nodes are interconnected with Intel® Omni Path Architecture (OPA) of 100 Gbps speed. The parallel file system is BeegFS with two servers. We compile the programs with GCC 8.3 and use Open MPI 3.1.4. Jobs have been submitted using slurm scheduler.
The code has been written in C programming language using MPI directives. mpiBWA encapsulates the original BWA-MEM version 7.15. Two implementations for mpiBWA exist: the first outputs one SAM file with all the chromosomes, the second (named mpiBWAByChr) outputs one SAM file per chromosome. Moreover, mpiBWA comes along with mpiBWAIdx, which is responsible for creating a binary image of the reference genome. This image is subsequently loaded in the shared memory by mpiBWA. Then, every mpiBWA process on the same computing node will share the same genome reference in order to save memory usage. mpiBWAIdx does not need MPI to run.
The Figure 5 shows the scalability of both mpiBWA (with the mpiBWAByChr implementation) and mpiSORT with varying sample sizes (from 28X to 300X) using computation distributed over 128 cores. The output files from mpiBWAByChr have been used as input by mpiSORT. Both algorithms efficiently scale as the input data are bigger, the walltime to process the data being proportional to their input size. We assessed how much time was spent on pure computation versus IO (i.e. reading the input files and writing the output files): mpiBWA spent more than 97% of its walltime in computation while mpiSORT spent between 50% to 60% in IO. The walltime to analyze the biggest sample of 300x is 9h hours for the alignment and less than one hour for the sorting.
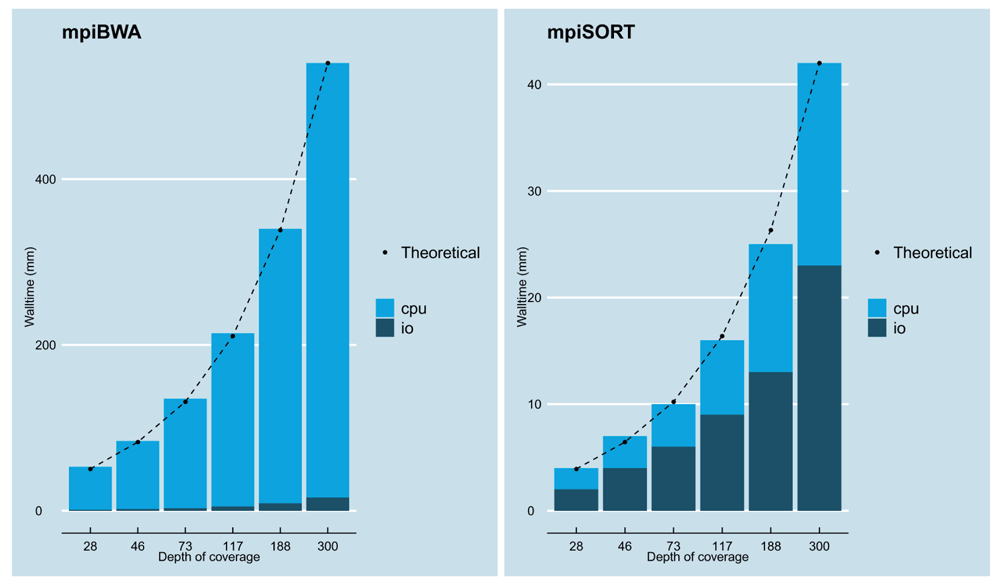
The FASTQ files are first aligned with mpiBWAByChr, SAM files are generated as outputs and then sorted with mpiSORT and -u option. 128 cores have been used over 16 and four nodes with mpiBWA and mpiSORT, respectively. The black dots represent the theoretical values of the walltime with respect to the 300X depth of coverage being set as the reference.
The first step consist in building the binary image of the reference genome that will be used in the shared memory by mpiBWA:
mpiBWAIdx hg19.small.fa
This creates the file myReferenceGenome.fa.map.
Importantly, mpiBWAIdx requires the following files that are generated by BWA index:
• myReferenceGenome.fa.sa • myReferenceGenome.fa.bwt • myReferenceGenome.fa.ann • myReferenceGenome.fa.pac • myReferenceGenome.fa.amb
The .map file needs only to be generated once. Then, mpiBWA is executed with the mpirun program, for example:
mpirun -n 2 mpiBWA mem -t 8 -o ./HCC1187C.sam hg19.small.fa \ HCC1187C_R1_10K.fastq \ HCC1187C_R2_10K.fastq
This command launches two processes MPI with eight BWA threads each (16 cores will be used). If you want to split the results of the alignment by chromosome, use mpiBWAByChr, for example:
mpirun -n 2 mpiBWAByChr mem -t 8 -o ./HCC1187C.sam hg19.small.fa \ HCC1187C_R1_10K.fastq \ HCC1187C_R2_10K.fastq
The total memory used during the alignment if approximately the size of the .map file plus the size of the SAM chunks loaded by each BWA tasks. A BWA thread takes around 300 MB of memory.
From the results obtained from mpiBWAByChr, each SAM files can be sorted as follows:
mpirun -n 4 mpiSORT -u chr1.sam ${HOME}/mpiSORTExample
This command launches four processes MPI.
As mpiSORT requires all the input SAM file to be loaded into the memory, the program is memory bounded. Therefore, a lot of attention has to be paid to define how many cores are needed to process the data. For example, let’s assume that the computing cluster consists of nodes with 190 GB of RAM memory with 40 cores each (thus 4.75 GB per core is available but this value can be rounded to the lower limit of 4.5 GB to leave some free memory on the node). To choose the number of cores, the following rule has to be applied: the total memory for sorting a SAM file that contains only one chromosome is around 2.5 times the size of the SAM file. For instance, sorting a chr1.sam file of size 209 GB with 4.5 GB per core requires 128 cores, for a of size 110 GB it requires 64 cores. We remind that the bitonic sort algorithm requires a number of cores that is a power of 2, this is the reason why the number of cores has to be set to the upper bound to the closest power of 2.
In this paper, we described parallel algorithms to process sequencing data that fully benefit from high-performance architecture (Hupé & Jarlier (2020a; 2020b)) that differ from other implementations using embarrassingly parallel approaches (Decap et al., 2015; Kawalia et al., 2015; Puckelwartz et al., 2014). Our implementation is based on bitonic sorting network, Bruck algorithm and IO optimizations with MPI directives. The scalability of the software strongly relies on the underlying hardware architecture, such as the low latency interconnection and distributed file system. Interestingly, our implementation allows the generation of aligned read files for each chromosome that can be further sorted in parallel thus reducing the time of downstream analysis whenever an analysis per chromosome is possible. The performance we obtained showed that MPI is very relevant for the field of genomics. The tools we developed pave the way towards the use of whole genome sequencing in daily clinics in order to meet the deadline and deliver results to the clinician in real-time for precision medicine as the time to delivery can be reduced to several minutes if the code is distributed over several hundreds of cores.
All data underlying the results are available as part of the article and no additional source data are required.
Source code for mpiBWA available at: https://github.com/bioinfo-pf-curie/mpiBWA.
Archived source code at time of publication: https://doi.org/10.5281/zenodo.3727143 (Hupé & Jarlier (2020a)).
Source code for mpiSORT available at: https://github.com/bioinfo-pf-curie/mpiSORT.
Archived source code at time of publication: https://doi.org/10.5281/zenodo.3727145 (Hupé & Jarlier (2020b)).
All documentation is available at: https://github.com/bioinfo-pf-curie/QUARTIC.
License: CeCILL Version 2.1.
F.J. and N.J. developed and tested mpiBWA. F.J., N.F., L.S., T.M, P.P. and M.F. developed and tested mpiSORT. M.M. provided technical expertise and access to computing cluster facilies to benchmark the code. F.J. coordinated the developments. F.J. and P.H. wrote the manuscript. P.H. supervised the study.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)