Keywords
non-tuberculous mycobacteria, NTM, taxonomic classification, MASH ANI, MLST
non-tuberculous mycobacteria, NTM, taxonomic classification, MASH ANI, MLST
The changes in the new version were made in response to the first reviewer. For a detailed description of the suggestions and comments and our responses, I will refer to the response to the reviewer we posted in responses, including descriptions for the changes in the manuscript, which are listed below:
Page 4: Abstract: Added “community standard” for MASH to clarify that the program does not originate with us, but instead is a widely used rapid genomic ANI calculator
Introduction: Made suggested syntax changes: italicized Mycobacterium as it is a genus name, removed a period, and added a comma.
Page 5: Results: Added first, second, third and finally, plus semi-colons to help separate the different facets and make the sentence more readable. Also added that the ANI mentioned in the second facet is related to the clustering in the first.
Page 7: Added clarification that disagreements in the NCBI names with our assignments are possibly due to divergent naming regimes and do not necessarily indicate different classifications.
Page 8: Added clarification that our genus assignments still do need organismal trait observations for full confirmation per a suggestion by the reviewer
Figure 1 Legend. The added sentence is to add similar clarification as in page 7.
Page 11: Added a period as suggested by the reviewer.
Page 12: We have added direct comparisons of our ANI and MLST results with Matsumoto et al (reference #44), specifically in Figure 1C, that were previously only in Supplementary Table S13. We have highlighted two divergent claims of the placement of individual genomes (M. avium) and of genus clades. We additionally have added a clause where the Matsumoto placement of a species confirms our placement.
See the authors' detailed response to the review by Charles Warden
Non-tuberculous mycobacteria (NTM) are a ubiquitous group of diverse environmental mycobacteria related to Mycobacterium tuberculosis, the bacterium that causes tuberculosis (TB)1–7. A significant number of NTMs cause clinical disease. Diseases caused by NTM infections are important sources of morbidity and mortality throughout the world, with a resurgence of infections in the United States, especially in immunocompromised individuals. The host-pathogen interactions of NTMs remain poorly characterized. Importantly, the background prevalence of NTMs has been shown to interfere with important aspects of TB disease control and management, including the efficacy of new TB vaccines8,9, established immuno-diagnostic methods, and the specificity of diagnostic and immune biomarkers10. Data on NTM prevalence in TB-endemic countries, however, is limited and often under-reported. Possible factors contributing to under-reporting include overlapping clinical manifestations of TB and NTM disease, lack of awareness of NTMs, and insufficient laboratory infrastructure to identify and culture NTMs11. The NTM-NET published a survey of NTM patient isolates12: “A total of 20 182 patients had NTM species cultured from pulmonary samples in these centres in 2008; 91.3% (n=18 418) of the isolates were identified to species/complex level; the remaining 1764 isolates (8.7%) were not identified beyond Mycobacterium species other than M. tuberculosis complex. A total of 91 different NTM species were encountered in this survey.” While this was an important survey of NTM, geographical distribution the low number of NTM species identified and the inability to classify some samples emphasizes the need for more classification resources. Gupta et al. proposed the Mycobacterium genus be divided into five distinct genera: Mycobacterium, Mycolicibacter gen, Mycolicibacterium, Mycolicibacillus, and Mycobacteroides13 in the Mycobacteriaceae family. There are over 197 Mycobacteriaceae species from these five genera according to the List of Prokaryotic Names with Standing in Nomenclature (LPSN)14,15 and elsewhere.
We undertook to sequence, analyze and characterize NTM strains representing geographically diverse TB-endemic areas with high NTM prevalence. Ancillary findings from adolescent and neonatal cohort studies conducted by Aeras and their collaborators to determine the incidence and prevalence of TB in Cambodia, India, Mozambique, South Africa, and Uganda included the prevalence and typing of NTM infections. The sites that participated in these studies collected NTM samples which were sequenced by JCVI. Extended data, Supplemental table S116 lists the samples sequenced from each site.
Our experience elucidated some of the issues which confound clinicians. Current commercial methods for typing NTMs are limited to a small number of the better known NTMs and even then are often not sensitive or specific enough2,17. Connecting the published literature for NTM type strains and related characterization of the species to a clinical isolate is often difficult even given the genome of the isolate due to non-specific marker gene sequences being available. This is being addressed to some extent as many more type strains are having their genomes sequenced18–20. We propose a fast average nucleotide identity (ANI) method when whole genome sequencing is practical and a multi locus sequence typing (MLST) approach when whole genome sequencing is not feasible.
There are four major facets of our analysis: first, clustering of Mycobactericeae family genomes using ANI; second, a pan-genome of representative genomes from each clade determined by ANI mentioned above; third, an evaluation of our MLST scheme; and finally, a comparison of our pan-genome to the work of Gupta in terms of genera assignment and genera specific genes. ANI-based clustering has been shown to be well correlated with known species and subspecies definitions21–30. We show how automated ANI clustering and ANI comparisons to representative genomes can inform classification of a novel genome. Pan-genome analysis can be used to evaluate genome relatedness across two major comparators: ANI as previously mentioned across all genes but also across subsets of genes, and gene content similarity. We discuss aspects of both pan-genome ANI and gene content. MLST can be used in the absence of genome sequencing data if the loci used in the MLST scheme are acquired by other methods such as PCR. We propose a distance metric based on the concatenated alignment of the MLST loci and evaluate how this can be used in the absence of ANI for classification. Gupta recently proposed a reclassification of five genera within the Mycobactericeae family13. We evaluate this proposal compared to our pan-genome analysis. Gupta also proposed genera specific genes to help delineate the genera and we evaluate those as well.
A total of 2,802 genome assemblies in the Mycobactericeae family in NCBI RefSeq were downloaded on 8 August 2019 (to reduce the number of genomes analyzed we only used complete M. tuberculosis genome assemblies). This represents the complete set as at 30 April 2019. We added six Mycobactericeae genome assemblies available in GenBank that had been excluded from RefSeq either for no specified reason or for being untrustworthy type strains, though the latter were not used to assign names to other genomes. An additional 11 genomes from other Corynebacteriales families that were sequenced as part of the project (Extended data, Supplemental table S116) were included in the analysis for a total of 2819 genomes. We generated species- and sub-species-level clusters of the Mycobactericeae genomes using an all versus all ANI comparison with MASH21,31. We generated species and sub-species clusters with average linkage and a cutoff value of 95% ANI for species and 98% for subspecies (Extended data, Supplemental tables S2 and S332) using GGRaSP33. This has been shown to be a reasonable way to computationally compute tentative species/subspecies clusters18,21–30. For the species-level 95% clustering, there were 269 clusters of which 263 are from Mycobactericeae, 126 of the Mycobactericeae clusters contained a type strain, of which 13 contained more than one type strain of different species (Extended data, Supplemental tables S2 and S334). For the subspecies-level 98% clustering, there were 336 clusters of which 329 are from Mycobactericeae, 137 of the Mycobactericeae clusters contained a type strain, of which 11 contained more than one type strain, and 1 which contained multiple subspecies (Extended data, Supplemental table S332). To represent as much of the taxonomic space as succinctly as possible, we selected representative genomes using clades generated from the two cluster levels. By default, we used the 95% ANI clusters, but for the nine 95% clusters that contained two or more 98% clusters containing type strains, we used all of the 98% clusters in place of the 95% clusters. This meant that 26 98% clusters replaced these nine 95% clusters, resulting in 280 Mycobacteriaceae clusters out of 286 total clusters, 131 of which contained a type strain, 12 of which contained more than one type strain of different subspecies (Extended data, Supplemental table S435). For each of the final 280 Mycobacteriaceae clusters, a representative genome was determined using GGRaSP based on the minimum of summed pairwise MASH ANI distances within the cluster which is called the cluster medoid genome. For the clusters with a RefSeq-included type strain, only the RefSeq-included type strains were considered for the representative genomes, whereas in the other clades, all genomes were considered.
We used the J. Craig Venter Institute (JCVI) pan-genome pipeline to generate a pan-genome using 279 of the 280 representative Mycobacteriaceae genomes plus a Norcadia genome (N. nova SAMN02900753, cluster 190) as an outgroup36,37. We were required to exclude one of the clusters (180B) as the sole cluster member (SAMN04216932) is not available in RefSeq, whose consistent annotation we use for the pan-genome pipeline. Using gene presence in at least 95% of genomes to determine core genes, we found 1,680 core genes (723 at 100% including the outgroup genome another 407 missing in one genome only) out of 118,867 orthologous gene clusters including 71,559 gene clusters present in only a single genome. A previously reported NTM pan-genome19 using only 99 genomes struggled to identify many core genes (243), possibly due in part to using a 100% threshold for genome presence and generated a lot of orthologous gene clusters (150,000), possibly due to the 80% nucleotide identity cutoff used. The pangenome results in multiple similarity measures which aid classification of the genomes. For example, identifying core genes is crucial for developing diagnostics, especially for identifying potential MLST loci with the least diversity to allow for primer design. Additionally, non-core genes which are restricted to only a segment of the phylogeny can be used for classification purposes, such as genus-specific genes for genus classification. Finally, the JCVI pan-genome pipeline produces pairwise genome ANI results based on the orthologous clusters determined from which genome ANI trees can be created.
We believe the ANI based clusters (Extended data, Supplemental table S435) and their representative genomes can be used for novel genome classification. How to classify clusters which contain more than one differently named type strain or contain no type strain at all is complicated. As Tortoli et al.18 point out, historical species definitions made before genome sequencing may need to be revised when existing species are found to have very similar genomes. Species should not be redefined simply based on ANI clustering since other factors such as significant difference in genome size (gene content), significantly divergent host specificity, disease presentation, or significant presence in the literature must be taken into account. We leave it to those more familiar with Mycobactericeae species to make those determinations. Three other problems were pointed out by Tortoli et al.18 which we also observed: some type strains have been genome sequenced more than once where the genomes are clearly different species from each other38; type strain genomes do not conform to species descriptions and previously sequenced marker gene sequences, and non-type strain genomes have been misnamed based on the first two problems. We identified two type strains: M. phlei (SAMEA4040484) and M. chelonae (SAMEA4040023) whose clustering with other type names suggests that their names have been swapped on submission to NBCI. We have used our corrected names for these samples in our tables and figures. As mentioned above, for clusters with more than one differently named type strain, we chose the medoid of the type strains as the cluster representative but in Extended data, Supplemental table S435 we give all of the type strains in the cluster and in Extended data, Supplemental table S539 we use an asterisk with the proposed species/subspecies classification name to indicate the uncertainty for members of these clusters. For species clusters lacking a type strain, a cluster-specific “genomosp.” (this is NCBI’s preferred designation for proposed species clades without a type strain) such as genomosp. X was used as the species name for all the genomes in the cluster. If a subspecies cluster did not have a type strain, then only the species level name is given since NCBI does not support a genomosp. concept at the subspecies level.
For all the Mycobacteriaceae genomes, we assigned a genus to them using average-linkage clustering based on PanOCT pan-genome pairwise ANI distances (Figure 1)36. For the four new genera which Gupta proposed, we found the least common ancestor node which contained all the Gupta genus type strains for that genus. Since Gupta supplied only one type strain for the Mycobacterium genus, we also found the root node of the largest subtree that included this type strain and no additional Gupta genus type strains. The genomes descending from these five nodes were then assigned to the respective Gupta genus. Other than the single outgroup genome not from Mycobacteriaceae, there is only one clade which is not included in the five Gupta subtrees. In Figure 1, this clade is indicated by black colored branches. By Gupta’s criteria for NTM genera within the Mycobacteriaceae this novel clade should be designated as a new genus. This clade contains four representative genomes but only one type strain, Mycobacterium haemophilum (SAMN01918491). We labeled these genomes as Unclassified genera in Extended data, Supplemental table S435, pending assignment of an additional genus name. NCBI has accepted the Gupta proposal in naming NTM species except for the misidentified genome, Mycolicibacterium vulneris (SAMN06651660)38,40. For this genome, NCBI has accepted the genus reassignment from the Gupta corrigendum, Mycobacterium vulneris which matches our clade assignment, though they have not renamed the species. Neither Gupta nor NCBI has renamed all NTM genomes in GenBank based on some form of clustering or phylogeny. This means the current NCBI names are not consistent with our clade-based assignment, as seen in the occasional differences in leaf and branch colors in Figure 1 and that these differences do not necessarily reveal divergent classification of the species using Gupta based genera. Of the representative type strains, four have NCBI genus names which disagree with our assignment. These are labeled as Mycobacterium (neumannii, syngnathidarumi, dioxanotrophicus and aquaticum), but are located in the Mycolicibacterium subtree. One of them (nuemannii) even shares a cluster with a Gupta Mycolicibacterium type strain (Mycolicibacterium flavescens). There are also 58 non-type representative genomes whose NCBI genus assignment disagrees with ours, and we have highlighted these rows in Extended data, Supplemental table S435. Our pan-genome ANI clustering was done for the representative genomes, so we conferred the same genus as determined for each representative genome to genomes in that representative’s species/subspecies cluster (Extended data, Supplemental table S539). This also identified four non-representative type strains which had a different genus than their NCBI name. All four were previously labeled as Mycobacterium, with two renamed as Mycobacteriodes (chelonae subsp. gwanake and subsp. chelonae), and two as Mycolicibacterium (kyoganese and lehmannii). However, these genus reassignments based on ANI should be confirmed with supporting growth and metabolic observations similar to the Gupta et al. results.
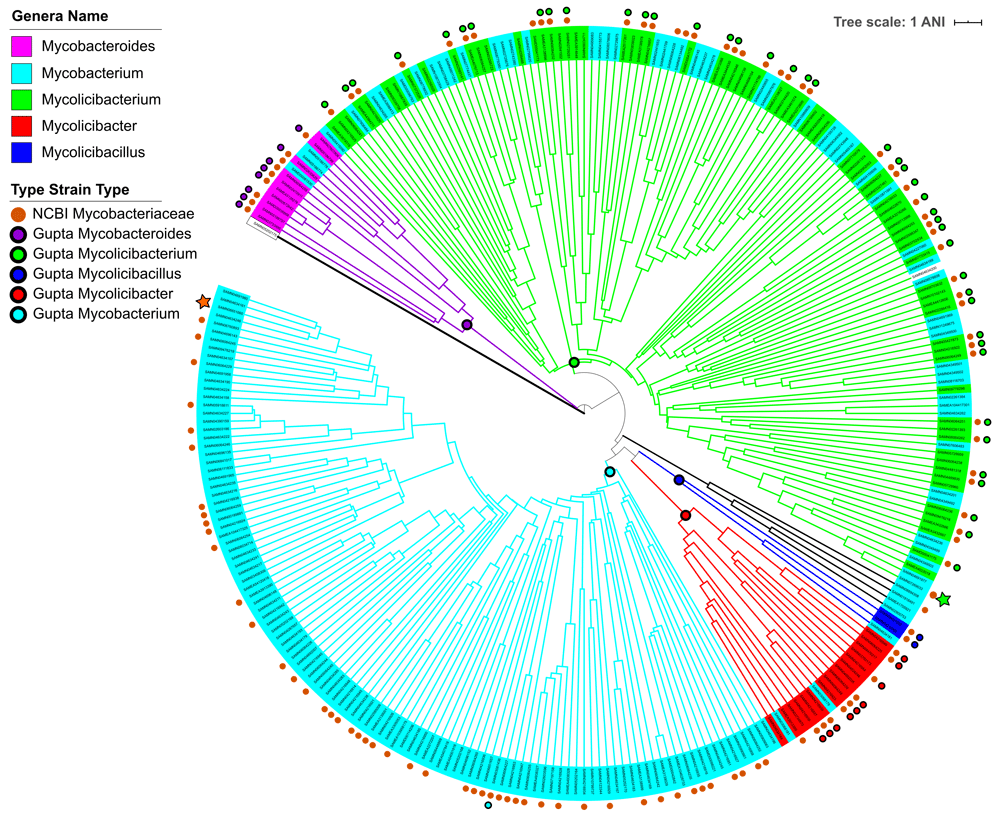
The tree was generated from the pan-genome ANI distance metric using average-linkage clustering. The branches are colored according to the assigned genera of the genomes from least-common-ancestors with the Gupta-defined type strains (shown on the tree as the colored circle on the ancestral nodes). The leaves are colored by the genus given by NCBI for the genome on 09/09/2019. Some of the NCBI genus names listed do not account for the Gupta et al. genus definitions, and thus branch and leaf color disagreements probably only reflect a difference in genus naming conventions. On the outside of the tree, the orange circles denote NCBI-defined type strains, whereas farther out circles indicate the type strains identified in Gupta et al. as belonging to a specified genus. The green star identifies the cluster containing two type strains for which the NCBI type strain genus identification and the Gupta assignment differs, and the orange star identifies the cluster containing a type strain previously suggested to be mis-identified.
The species and sub-species classification of all the non-representative genomes was done based on cluster membership. For genomes within clusters containing a single named type strain, that species name was used for all genomes. For clusters lacking a type strain, a cluster-specific “genomosp.” was used as the species name for all the genomes in the clade. For clades with multiple named type strains, the species name of the best ANI match to one of those type strains was used. We warn that the above caution about assigning species based solely on ANI holds and that further work might be necessary. The classification results are shown in Extended data, Supplemental table S539.
We propose classifying a novel genome by running MASH ANI of the novel genome compared to the set of representative genomes. To verify this approach, we classified each genome using MASH pairwise ANI to the set of representative genomes compared against the cluster-based naming. For each genome the closest MASH ANI to the representative genomes were almost exclusively within the same cluster. Only for 26 genomes in clusters 28B as well as single genomes in cluster 47B and in cluster 165B was the closest ANI assigned match not in the same cluster. This only occurred for clusters discriminated at the 98% threshold (subspecies). For the 26 genomes in cluster 28B but with a best ANI match to the cluster 28A’s representative genome, clusters 28B and 28A are two of the four subspecies clusters of M. abscessus. There are only three named M. abscessus subspecies and the fourth cluster (28A) is probably artifactual occurring right at the boundary of the 98% MASH ANI threshold. Clusters 28A and 28B should probably be combined based on the simple observation that all genomes in both clusters had their second-best MASH ANI match to the other’s representative sequence and all these ANI distances were less than 2% which is within the clustering threshold. The same is basically true for clusters 47B/E and 165B/A where only a few second-best MASH ANI distance values are slightly above 2%. Regardless of whether these clusters are merged based on these criteria when a novel genome is being evaluated the second-best MASH ANI distance should also be considered to ascertain the certainty of an assignment. Any novel genome exceeding the 95/98% ANI threshold to this set of genomes can be assigned a species/subspecies name using MASH ANI to representative genomes (again with more careful scrutiny if the second-best MASH ANI distance is very close to the best MASH ANI distance). The set of MASH sketches will need to be updated both as more genomes are sequenced falling outside of the existing clusters and as real species/subspecies names are defined or determined. While recent NTM type strain sequencing projects (BioProjects: PRJNA35424818, PRJDB422720, PRJNA29946719) have greatly increased the number of sequenced NTM type genomes, many have still not been sequenced and could correspond to some of the proposed genomospecies. Genomes sequenced as part of our project (Extended data, Supplemental table S116) have assisted in the expansion, contributing genomes to 88 of the 280 Mycobacteriaceae clades. This includes 58 clades for which they are the only representatives and a third of both the Mycobacterium and the Mycolicibacullus clades.
One common diagnostic tool used for identifying species or strains is multi-locus sequence typing (MLST), where a small set of single copy conserved gene sequences are used to distinguish genotype. MLST can be done computationally using genome sequences or via primer-based sequencing. In either case it is important that the gene sequences be conserved enough to design primers or find the gene via homology search; and divergent enough to be able to distinguish different species or strains. We found that some marker genes that had been used in the past41 tended to be either fragmented in genome assemblies or too divergent to align easily. When working with an earlier pan-genome we chose the core genes with no variation in gene length and highest conservation to serve as marker genes which all turned out to be ribosomal proteins (S7, S8, S12, S14Z, S19, L16, L19, L35). This MLST schema was submitted to PubMLST Mycobacteria42. These eight marker genes are also core genes in the 280 Mycobacteriaceae genome pan-genome with very low gene length variation and high conservation (Extended data, Supplemental file S634).
When genome sequence is not available for a clinical sample but MLST is a possibility, we believe that our or a comparable MLST schema could be used for diagnostic species/subspecies assignment, such as seen in Extended data, Supplemental table S743. Using our MLST schema an ST is never shared across species/subspecies clusters (Extended data, Supplemental table S539). For most of the genomes, the ST distance (the fraction of mismatches across the combined multiple sequence alignment of the MLST loci) of a genome to the representative genome of that genomes ANI cluster is smaller than the ST distance to any other representative genome (Extended data, Supplemental table S539). Only for 142 genomes was the closest ST different than the ANI assigned cluster (Extended data, Supplemental table S539). These genomes include 27 out of the 28 genomes whose best MASH hit to a representative were not in the same cluster as themselves, excluding only SAMN07811434 from Cluster 165B. This difference mostly occurred for an ST when the incorrectly chosen ST was from a different subspecies cluster of the same species, though for 59 genomes they were from different species clusters. For the 59 genomes which had best ST hits to different species the ST distance was above the cutoff for same species assignment explained below. For almost all 142 discrepant genomes the second closest ST was the correct one, with the exception of four cluster 47E genomes where the best ST hit was to 47D, and the second-best ST hit was to 47B. A remaining issue is whether a novel genome is within an existing cluster or a novel cluster. Figure 2 shows statistics for ST distances versus species and subspecies ANI ranges. We can see from this data that ST distance for 95–98% ANI ranges from 0.004–0.02, 98–100% ANI from 0–0.01, and <95% ANI from 0.006–0.26. We devised cutoffs to separate out the three groups using two standard deviations from the mean ST distance for 95–98% ANI (0.007 ± 0.003) which separated the three ANI-based groups (Different Species > 0.01 ≥ Same Species ≥ 0.004 > Same Subspecies) with minimal misassignment (Figure 2). Diagnosticians should use this data to inform the likelihood that a novel ST is from a known or unknown species/subspecies cluster.
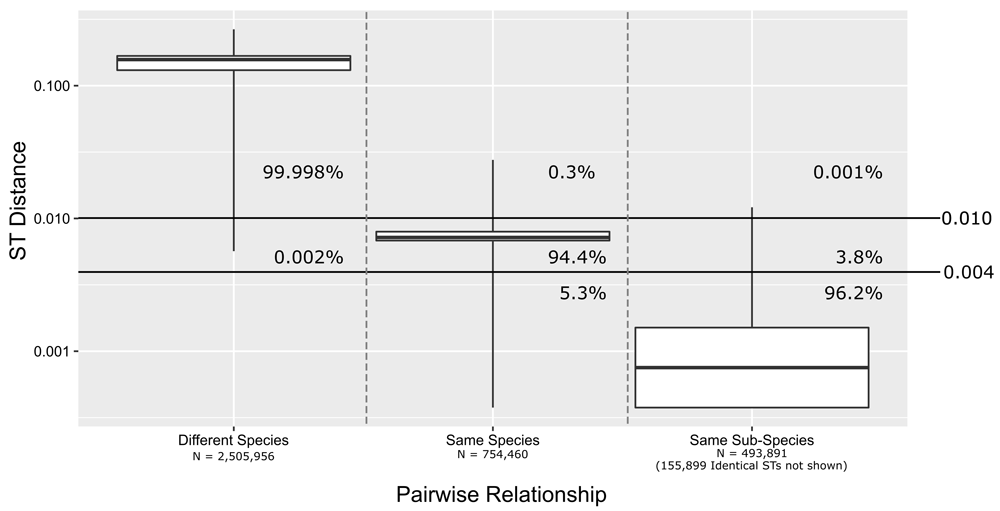
The distribution of ST distances between pairs of Mycobacteriaceae genomes classified by ANI clustering. Three categories of pairwise relationships were examined: genomes of different species (MASH ANI < 95%), genomes of the same species (95% < MASH ANI < 98%), and genomes of the same sub-species (MASH ANI > 98%). ST distance cutoffs to divide the groups were calculated from the mean distances of genomes from the same species plus or minus two standard deviation (solid horizontal lines). These successfully classified nearly all of the data. The percentage of each category of ANI matches which fall into the ST distance bins given by the two thresholds (0.01 and 0.004) are shown directly on the figure.
A recent article published after our analysis had been completed describes what is called an MLST approach to determine NTM species/subspecies classification44. Matsumoto et al. do compare their method against several others including our pubMLST scheme. They do not use our LOCUST software and it is not clear exactly which software on the pubMLST website they do use to evaluate our MLST scheme. For their 29 test genomes, they find that our MLST scheme correctly assigns all genomes at the species level but does not give a subspecies classification as theirs does. This makes sense because the profile we uploaded to pubMLST at the time of their analysis did not include subspecies level information. We have recently updated our pubMLST profile to include subspecies information. The MLST scheme presented by Matsumoto et al. is atypical. First, the set of loci used is very large (184), which is not consistent with performing efficient PCR. Second, only 53 of the loci are core genes; whereas, the remaining 131 are species specific genes. Third, because most of the loci are species specific it is not even clear how one defines a sequence type and what the authors discuss instead is a similarity score. Fourth, the msltverse software which is used (and is not compatible with standard MLST schemes) needs whole-genome read data as input obviating any advantage of MLST over whole-genome sequencing. As we have argued, whole-genome data for ANI comparison is preferable to an MLST approach but we also argue that the ANI data is sufficient for species categorization and that mlstverse is not necessary. We have nothing against looking for the presence or absence of genes in whole-genome sequence data to help identify virulence or antibiotic resistance. We do caution that only WGS data which can be assembled into complete chromosomes and plasmids can reliably determine the absence of a gene. We also note that claiming a particular gene is species/subspecies specific is only possible if a large, diverse set of genomes is available for all species/subspecies. Finally, attempting to do species/subspecies classification based on gene content is problematic due to horizontal transfer, particularly across subspecies boundaries.
To further aid in the genus classification of novel Mycobacteriaceae genomes, it is also possible to look for the presence of conserved, genus-specific genes. Gupta et al. provide 89 conserved signature proteins which can be used to define the family and genera (Extended data, Supplemental table S845)13. These proteins were selected by Gupta et al. based on BlastP comparisons of the genera type strain genomes and either the non-Mycobacteriaceae protein databases or Mycobacteriaceae genomes outside of a given genus Using our Mycobacteriaceae pan-genome with the genus assignments discussed above, we identified genus specific gene clusters which were present in each representative genome in a genus but not in any of the other genomes. If no cluster met this criterion for a genus, we required the difference between a gene clusters presence in the genus genomes and the out of genus genomes to be greater than 90%. We used the medoids of these selected gene clusters as potential CSPs. This resulted in 648 pan-genome based CSPs (Extended data, Supplemental table S945). Only a small number25 of the Gutpa CSPs were mapped to the pan-genome’s CSP clusters using either GenBank identifier matches or best blast matches probably due to different criteria for selection. Most46 of the remaining 65 of the Gupta et al. CSPs were genus-specific, but only had a partial presence in the genomes associated with the genus.
To test the genus prediction success rate of BlastX searches of these gene sets, we compared them to the 2,802 RefSeq Mycobacteriaceae genomic sequences. We did not include 11 of the Gupta CSPs which have since been removed from RefSeq (Extended data, Supplemental table S845). Classification of the genomes with the CSPs was based on a simplified assignment algorithm, where a genome was assigned to the genus with the greatest percentage of genus-specific genes that had a blast hit to a given genome with an e-value < 1e-5. The Gupta CSPs gave the same answer as the pan-genome based genus assignment for all the RefSeq Mycobatericeae genomes excluding the Unclassified clade 98.7% of the time, whereas the pan-genome CSPs exceeds this with a 99.9% match rate, even when accounting for the Unclassified genus. The one incorrect assignment was an Unclassified genome being assigned as Mycobacteroides. The assignment could potentially be improved by expanding the search beyond only a single medoid gene for each gene cluster. Overall using the CSPs to assign genus seems to have limited utility compared to MASH ANI or MLST. Researching why certain genes are genus specific and what phenotype(s) they confer might be of more value.
After our analysis was complete, some novel high-quality NTM genomes became available from Matsumoto et al.44: “In this work, we expanded the NTM genomic data by 63 species, of which 27 species were newly determined and the remaining 36 species were re-sequenced by Illumina and Oxford Nanopore Technologies (ONT).” We compared these 63 genomes along with the genomic classifications in NCBI and the species tree in Figure 1C in Matsumoto et al.44 to our cluster representatives using ANI and ST distance (Extended data, Supplemental table S1347). The classification of the genomes using these strategies and the mlstverse reported in Matsumoto are highly similar, though there were differences. Two dis-similarities include the imposition of the Mycolicibacillus and Mycobacteriodes genome clades into the larger Mycolicibacterium clade in the Matsumoto et al. Figure 1C44 as compared to the fully distinct clades in the pangenome tree (Figure 1), and the different placements of M. avium in the two trees, with the pangenome placement more reflective of earlier suggestions and the genera name. We also used our and Gupta’s CSP proteins to predict genera (Extended data, Supplemental table S1347). Of the genomes, 16 are not in any of the clusters by ANI (95% threshold). Of these genomes, 15 currently have genus names that agree with their closest MASH medoid (four Mycobacterium and eleven Mycolicibacterium). The single genus name disagreement is BioSample SAMD00153170, which is currently labeled Mycobacterium gallinarum, but which is closest to a Mycolicibacterium clade with a MASH distance well within the generally accepted ANI range (94.19%) of being in the same genus and is further located in a Mycolicibactierium clade in the tree shown Matsumoto et al. Figure 1C44. Additionally, seven genomes are in previously untyped clusters, thus enabling the clades to be named with a species name (Extended data, Supplemental table S435). For 47 of the 63 genomes the best ANI match to a cluster representative is greater than 95%. Of these 47, 33 have the same species name as the representative, 7 as noted previously do not have a real species name, and 7 have different species names. Mycobacteroides abscessus subsp. massiliense has its best ANI match to cluster 28A which is one of the four subspecies clusters for Mycobacteroides abscessus but the previously sequenced type strain for Mycobacteroides abscessus subsp. massiliense is in cluster 28B. We had already observed that clusters 28A and 28B should probably be merged based on ANI and ST mapping and this only reinforces that observation. For the 7 with different species names, there are four categories: split the existing cluster into two or more clusters at 98% ANI to represent different species (three genomes with 95.1–96% ANI to cluster representative), split the existing cluster into two or more clusters at 98% ANI to represent different subspecies (two genomes with 97.6–98.3% ANI to cluster representative), consider declaring two type species to be the same species and one of them to be a heterotypic synonym (1 genome with 99.0% ANI to cluster representative), and the genome type strain is already in the cluster but the chosen medoid between the two species is not the same (paraintracellulare and intracellulare in cluster 47D). This leaves 16 genomes with a best match to a cluster representative below 95% ANI. These 16 genomes would found new clusters where each would be a singleton cluster and be the representative for that cluster. Of the 63 genomes 25 had novel ST types which we added to the pubMLST schema. The ST distances were generally well correlated with the ANI values with only 4 genomes with ANI below 96.1% having ST distances above the same species threshold. For the CSPs neither Gupta (five discrepant) nor ours (one discrepant) performed perfectly for genus assignment. Mycolicibacterium aubagnense was classified by both methods as Mycobacterium, and four additional genomes had discrepant Gupta CSP based genus assignments. This confirms our skepticism about this approach.
Our analysis shows that the universe of NTM species/subspecies is still far from complete: 142 of our clusters do not even have an associated type strain, many NTM species still do not have a genome sequence, and there are probably many NTM clades which are not represented in our clusters. Our suspicion of the sparseness of the NTM family tree is reinforced with the addition of the Matsumoto genomes, a quarter of which were not assigned to our species-level clusters. The incompleteness of NTM genomic data could easily lead to misdiagnosed or undiagnosed NTM infections. We have proposed an efficient structure based on ANI and species/subspecies cluster representative genomes to classify any patient sample with genomic data. This includes recognizing that a patient sample is from a novel NTM clade. As more NTM genomic data becomes available, our representation is easily extended with new species/subspecies cluster representative genomes. Many NTM species are slow or very slow growing making isolation of DNA for genomic sequencing challenging. When genomic data is not available, we show that our pubMLST scheme is sufficient for the classification of most patient samples. Diagnostic companies may not prefer our choice of MLST loci, so we have included all of our data on the pan-genome clusters (Extended data, Supplemental file S634) from which to choose for PCR or hybridization-based methods. We chose our loci based on being more highly conserved at the nucleotide level for PCR primer design, being present in all genomes, being full length in all genomes, and having low length variability so that gaps would not complicate multiple sequence alignment-based analysis (Extended data, Supplemental file S634). When more extensive analysis is desired, for example for the presence of noncore antibiotic or virulence genes a pan-genome analysis can be helpful. We showed that the pan-genome can be used to determine core and genus-specific genes.
A total of 288 samples of genomic DNA were collected from five different geographical sites (Extended data, Supplemental Table S116). The genomic DNA was isolated from cultures grown from samples of sputum or gastric aspirates that were originally collected from TB presumptive patients with the exception of an adolescent cohort study (E-004-IN-A) from the St. John’s Research Institute (SJRI), where the specimens were collected from healthy children. Samples were initially treated with a decontamination and digestion step using N-acetyl-L-cysteine-sodium hydroxide (NALC-NaOH) and then screened for acid-fast bacilli (AFB) using smear microscopy with Ziehl-Neelsen (ZN) or Auramine O fluorescent staining (South African Tuberculosis Vaccine Initiative (SATVI) and Makerere University (MU). Samples obtained from SJRI, Centro de investigação de Saúde de Manhiça (CISM), and MU were also examined directly by ZN and fluorescent microscopy prior to decontamination. After decontamination and digestion, samples were cultured on Lowenstein Jensen (LJ) media and also inoculated into Mycobacterial growth indicator tubes (MGIT) 960 BD, following manufacturer’s instructions. Samples from SATVI that were MGIT positive for growth were then stained for AFB by ZN and Gram stain. Samples from all sites that were AFB-positive were further identified at the genus and species levels. Samples at Institut Pastuer – Cambodia (IPC) and SATVI were initially analyzed by AccuProbe, Gen-probe, Inc., while samples at MU were first typed by mycobacteria specific 16S rDNA PCR identification. All sites, with the exception of SATVI, used the Hain GenoType Mycobacterium CM/AM test system, according to manufacturer’s instructions, to identify NTMs at the species level. Samples at SATVI that were Accuprobe negative were further identified by methods of Brunello et al.48 and Telenti et al.49 (Briefly, PCR amplification of hsp65 gene, digestion of product by 2–3 restriction enzymes, identification by RFLP – examination of banding patterns on agarose gel electrophoresis); see Hatherill et al.50 for further details.) If any other bacteria were detected during smear microscopy examination for AFB, the cultures were subject to a second round of NACL-NaOH decontamination and reinoculated into LJ and MGIT. The contaminated cultures were also plated onto blood agar to identify contaminants. All decontaminated specimens positive for AFB were saved and stored at either -20°C (CISM) or -80°C (IPC, SJRI, MU, SATVI). Positive growth cultures from the MGIT were also stored at -80°C in the MGIT media of Middlebrook 7H9.
In an attempt to provide broad coverage of the NTM strains both by species and geographic location, no more than 10 strains of the most frequent species from each site were chosen for regrowth. At the same time, no fewer than five strains from the same species were collected when possible. Additionally, a number of unidentifiable NTM cultures were also collected from each of the sites. Samples within these parameters were chosen at random for regrowth; however, samples that failed to regrow were replaced with new samples. Sites noted that samples frozen at more recent dates were easier to regrow. Regrowth of cultures was done from MGIT Middlebrook 7H9 or decontaminated specimen frozen stocks by inoculating MGIT 7H9 media (CISM, IPC, MU, SJRI), LJ medium (CISM, SATVI/CSU), and 7H11 (SATVI/CSU, CISM). Subculturing on LJ media for single colony isolation was done for samples from CISM, IPC, SATVI/CSU, and SJRI. Regrown samples from MU were observed by ZN staining to re-confirm presence of AFB and also rule out contaminants by culturing on blood agar.
NTM cultures were grown in 7H9 medium up to an OD >0.6 and then centrifuged for five minutes at 10,000 rpm. Supernatant was removed and pellets resuspended in TE buffer before incubating at 95°C for 10 minutes. Several sites grew cultures on solid media instead of 7H9 prior to DNA extraction. This media included 7H11 agar (SATVI) and LJ agar (SJRI, CISM, CISM). For these samples, single colonies were used for DNA extraction in TE buffer. After the incubation step, samples were centrifuged for 10 minutes at 10,000 rpm and supernatant removed and passed through a sterile syringe Filter (0.2 µm). Samples were stored at 4°C.
Illumina NextSeq library preparation and sequencing was used. Genome assembly was performed using SPAdes v 3.1.151, which produced good quality assemblies as detailed in supplementary materials (Extended data, Supplemental Table S116).
The NTM genomes sequenced for this project were annotated by NCBI’s prokaryotic annotation pipeline, PGAP52.
A total of 2,802 NTM and TB complex strains were downloaded from NCBI RefSeq on 08/08/2019 and their PGAAP annotation used for comparative analysis. 6 Mycobactericeae strains excluded from RefSeq for either being untrustworthy as type or for no given reason and 11 Cornybactereae sequenced by as part of this project were also downloaded. ANI between genomes was calculated using MASH v2.0, a very fast tool for determining approximate pairwise ANI values based minHashing of kmer overlap31. Pairwise comparisons of all the genomes was run with 10,000 21-mers per genome. We used the GGRaSP R package v1.0 which generated a complete linkage tree from the distance matrix calculated by subtracting 100 from the MASH ANI results33. The resulting tree was divided into clusters at 95% (species level) and 98% (sub-species) ANI cutoffs using R commands ggrasp.cluster(threshold =2) and ggrasp.cluster(threshold =5). For 95% ANI clusters which contains at least two daughter 98% clusters with species or sub-species type strains, all the 98% clusters are retained; otherwise, only the 95% clusters are retained. All clusters are named by the GGRaSP 95% numeric order, and the 98% cluster having a sequential additional letter. For each cluster, a representative was selected by picking the type strain that has the lowest combined ANI to all the other genomes in the cluster. For clusters with no type strain, the genome with the lowest combined ANI to all the other genomes was selected. If a cluster contained a single type strain or a Matsumoto genome was later assigned to a cluster with no type strain, then genomes in that cluster were named based on that type strain. For clusters with multiple type strains genomes were also individually named from MASH ANI comparisons with type strains (both those downloaded on 8/8/2019 and those subsequently sequenced by Matsomoto) using the name deriving from the type strain with the highest ANI value. Species names are only used when the ANI value is greater than or equal to 95% and sub-species for values greater than or equal to 98%.
We used the JVCI pan-genome pipeline (default settings using nucleotide sequences) to generate a pan-genome using 279 of the 280 representative Mycobacteriaceae genomes plus a Norcadia genome (N. nova SAMN02900753, cluster 190) as an outgroup36,37. We were required to exclude one of the clusters (180B) as the sole cluster member (SAMN04216932) is not available in RefSeq whose consistent annotation we use for the pan-genome pipeline. When representative assemblies were not available in RefSeq (two clusters: 141, 188A), but assemblies from other genomes within the cluster were, a secondary representative present in RefSeq was used.
The genus of the 279 mycobacteriaceae medoiods was assigned using the pan-genome ANI average-linkage clustering generated from the JCVI pan-genome pipeline36,37. For four of the genera, the Least-Common Ancestor for all genus type strains designated by Gupta et al. (found either within the text or in Tables 8–11) was identified13. Since the genus Mycobaceterium only had a single designated genus type strain, the most ancestral node containing the genus type strain and no other type strains was used as the genus ancestral node. All genomes descending from these nodes were assigned to that genus. Any remaining Mycobacteriaceae genomes were in a single cluster and labeled as unclassified. All non-representative genomes were assigned the same genus as the representative strain in their cluster.
Core genes identified by pan-genome analysis36,37 were evaluated to identify candidate marker genes for MLST analysis. Eight highly conserved ribosomal proteins (S7, S8, S12, S14Z, S19, L16, L19, L35) were selected. Distinct allelic profiles and sequence type definitions were generated by searching the consensus sequences of the eight ribosomal proteins against 2,802 NTM and TB complex strains using the novel schema function of LOCUST v3.053. The corresponding allele sequences and full MLST schema was submitted to https://pubmlst.org/mycobacteria/. Species and subspecies assignments were made for 2,802 NTM and TB complex strains using the cluster approximation function of LOCUST version 3.0 (manuscript in preparation) based on the 280 type and proxytype strains discussed previously (Extended data, Supplemental table S435) with the command line typer.pl –biosample_list biosample.list –download_schema “Mycobacteria” –cluster_approximation –cluster_approximation_list type_strain.list -t fasttree
The programs used in this study are available through GitHub at the JCVI repository at https://github.com/orgs/JCVenterInstitute, including: JCVI pan-genome pipeline (PangenomePipeline), LOCUST, and GGRaSP.
Genus Conserved and Unique Proteins were selected for each of the six Mycobacteriaceae genera by identifying clusters which are present in all the genomes of a genus and absent in all other genomes. When there were no such clusters for a genus, clusters where the percentage difference between presence in the genus and presence outside the genus was greater than 90% were selected. The medoids for each of the selected clusters were used. Protein sequences of the cluster centroids were obtained using transeq in the EMBOSS suite. Protein identifications for all of the Gupta et al. genes were collected from tables 2, 5, and 713 and the sequences were downloaded from NCBI. Comparisons of the presence and absence of the CSPs in the genomes was done using tblastn for the whole genome sequences and the CSPs. Blast matches with e-values < 1e-5 were considered a hit.
NCBI BioProject: Sequence and Analysis of Non-tuberculous Mycobacteria (NTM) strains. Accession number PRJNA305922.
This BioProject contains all clinical NTM sample sequences; Individual biosample IDs of the genomes can be found in Extended data, Supplemental table 116.
MLST allele sequences and corresponding schema is available at the PubMLST Mycobacteria genome database (https://pubmlst.org/mycobacteria/). All MLST profiles generated as part of these analysis contain the specific genome names in the organism column in Extended data, Supplemental table 743. Other MLST types have NA in the column.
Figshare: Supplemental Table 1. JCVI NTM Genomes. https://doi.org/10.6084/m9.figshare.12016290.v116.
Figshare: Supplemental Tables 2 & 3: NTM Genomes MASH ANI-based Clusters. https://doi.org/10.6084/m9.figshare.12024051.v132.
Figshare: Supplemental Table 4: NTM Clusters. https://doi.org/10.6084/m9.figshare.12024057.v135.
Figshare: Supplemental Table 5. NTM Genomes. https://doi.org/10.6084/m9.figshare.12024117.v139.
Figshare: Supplemental File 6. Pangenome Matchtable. https://doi.org/10.6084/m9.figshare.12024126.v134.
Figshare: Supplemental Table 7. NTM ST Definitions. https://doi.org/10.6084/m9.figshare.12024144.v143.
Figshare: Supplemental Tables 8 and 9. Analysis of Gupta et al. 2018 and Pangenome-Derived Conserved Signature Proteins in NTM Genomes. https://doi.org/10.6084/m9.figshare.12024159.v245.
Figshare: Supplemental Table 10: NTM Pangenome-Based ANI All Versus All Table. https://doi.org/10.6084/m9.figshare.12024024.v254.
Figshare: Supplemental Table 11: NTM Genome MASH ANI Distances. https://doi.org/10.6084/m9.figshare.12024030.v246.
Figshare: Supplemental Table 12. NTM All versus All ST-based Distance Matrix. https://doi.org/10.6084/m9.figshare.12024303.v155.
Figshare: Supplemental Table 13. Analysis of Genomes published in Matsumoto et al. 2019. https://doi.org/10.6084/m9.figshare.12024324.v147.
Extended data are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)