Keywords
Cancer cachexia, anoxia, intracellular lactic acidosis, weight loss, nutritional supplements, quality of life, Cori cycle, dichoroacetate, emodin, pentoxifylline
Introduction
Cancer cachexia (CC), also known as cachexia-anorexia syndrome1, is a consequence of cancer in which patients lose weight with an overall decline in health2. It is a combination of starvation and metabolic disturbances3 that greatly affects quality of life disrupting regular daily activities. In the more advanced states, cancer anorexia-cachexia cannot be modified or improved by increased feeding, stimulating appetite, or nutritional supplements4.
The cardinal clinical points of this syndrome are5:
Progressive and relentless weight loss (more than 5% of loss compared with normal weight).
Loss of muscle mass.
Loss of fat tissue.
Minimal or no response at all to usual therapies such as nutritional supplementation.
Difficulties in routine daily activities.
Marked fatigue or asthenia.
Loss of appetite.
Progressive deterioration.
Reduced cancer treatment tolerance.
Reduced length of life. (There is a correlation between weight loss, rate of weight loss and survival.)
Anemia.
The reduction in food intake alone does not explain the metabolic changes found.
Resting energy expenditure (basal metabolic rate) is increased in some cancers but not in others that also produce cachexia.
Associated with insulin resistance.
Associated with acute-phase inflammatory reactants.
In advanced phases of cancer up to 80% of patients exhibit CC6,7. Complex associations between cancer, host, nutrition, psychological, systemic and environmental factors were thoroughly studied as part of the problem. However, a unified and physiopathological explanation is lacking. This led to consideration of CC as a multifactorial consequence of cancer. Based on the thousands of publications and findings on CC, it is the objective of this work to arrange the multiple pieces of evidence published in the world medical literature and to build a comprehensive and unified picture of the syndrome. Understanding how anoxia, rather than hypoxia, is the main culprit of CC will allow for a different approach to treatment on a rational basis. A certain amount of speculation is involved in the proposed solution of the puzzle; however, this speculation has been kept to a minimum. Only laboratory work and clinical trials can ultimately confirm the validity of the hypothesis developed here.
Known mechanisms of cancer cachexia
As already mentioned, CC is described as the consequence of cancer-induced loss of appetite (reduced food intake-starvation) and metabolic alterations. It is found frequently in advanced tumors and many authors consider it a paraneoplastic syndrome8. Many mechanisms contribute to unleashing cancer cachexia. Some are known, others are not. However, a superficial look may be misleading because the problem is far more complex than it may seem. There are well known factors that play a role, which are summarized below.
Lack of appetite: reduced food intake
Although this may seem to be the main cause, it is not. Pharmacologically improving the appetite or increasing food intake does not solve the problem. Usually, it does not stop progressive weight loss. Anorexia is frequent, but there are many patients who lose weight without a manifest loss of appetite. Important contributory factors to anorexia are depression9,10, and cancer treatments themselves, such as chemotherapy and radiotherapy11,12. However, this treatment-related weight loss does not seem directly associated with cancer cachexia.
Inflammatory and catabolic mediators
These mediators were found increased in cachexia, such as tumor necrosis factor alpha (TNFα)13–15, ZAG (Zinc-α2-glycoprotein also known as lipid mobilizing factor)16,17, interleukin (IL)-118, IL-619, IL-1520, proteolysis inducing factor (PIF)21, myostatin22, and transforming growth factor-β (TGF-β)23, among others. Levels of glucocorticoids are also increased. These mediators seem to be part of the problem but not the cause. For example, TNFα and IL-6 produce loss of appetite by interacting with hypothalamic receptors that regulate food intake24. However, steep elevation of IL-6 is found mainly in very advanced stages of CC25. IL-6 is also involved in an autophagy inducing activity found in serum of patients with CC. Blocking IL-6, this activity disappears26.
Increased energy expenditure (higher basal metabolic rate)
A previous study showed that 143 out of 297 (48%) unselected cancer patients exhibited increased resting energy expenditure27. Several authors have confirmed increased energy expenditure as a cause of weight loss in cancer patients28–33. This probably is the consequence of increased uncoupling at the electron transport chain and increased Cori cycle activity. In the liver, the excess lactic acid produced by the tumor can be converted into glucose (Cori cycle) consuming ATP. The Cori cycle is considered an important culprit in cachexia. The Cori cycle (also known as the lactic acid cycle), converts lactate produced by anaerobic glycolysis in muscles to glucose in the liver. In cancer, instead of muscle, the tumor is the provider of lactic acid. While glycolysis produces a positive balance of two molecules of ATPs, the Cori cycle uses up six molecules of ATPs. Each turn of the cycle represents a net loss of four ATP molecules34. The Cori cycle has been held responsible for energy loss in cancer by many authors35–43. An intracellular Cori cycle cannot be ruled out as the main cause of energy loss (Figure 1).
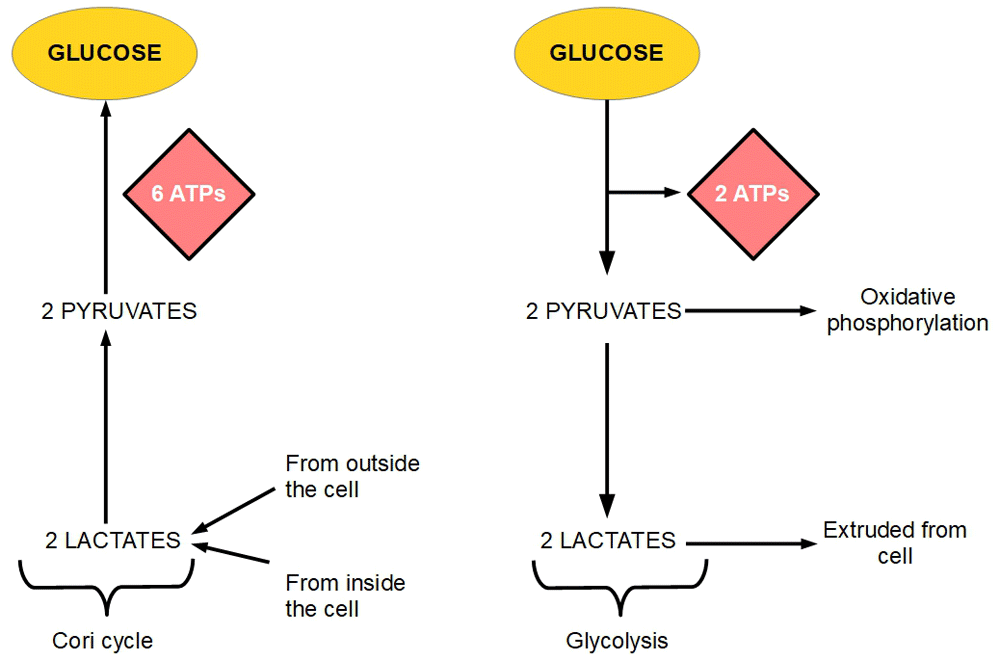
Figure 1. The Cori cycle and glycolysis.
The figure shows that six ATP molecules are necessary to convert two molecules of lactate into glucose (left panel). On the other hand, the glycolytic pathway (right panel) produces two ATP molecules by degrading glucose to lactate. If two molecules of lactate produced through glycolysis are reconverted into glucose, there is a net loss of four ATP molecules. If this is established as a permanent circuit glucose-lactate-glucose circuit, each turn in the circuit loses four ATP molecules.
Interestingly, the Cori cycle is activated during fasting44 where it contributes to generating glucose. Patients with advanced metastatic cancer show an increased Cori cycle, particularly those patients with high glycolytic flux35,36,45,46.
According to our criteria, the Cori cycle by itself, is the main responsible actor in CC, but not the originating cause. Figure 2 and Figure 3 show a proposed mechanism of how the Cori cycle develops in cancer and the energy imbalance it drives. For each molecule of glucose produced through the Cori cycle, six molecules of ATP are used. For each molecule of glucose degraded to lactic acid only two molecules of ATP are produced. Therefore, if a cycle is established in which one molecule of glucose produces two ATP molecules, and the lactic acid thus formed is used to regenerate glucose, four molecules of ATP are lost in each complete cycle.
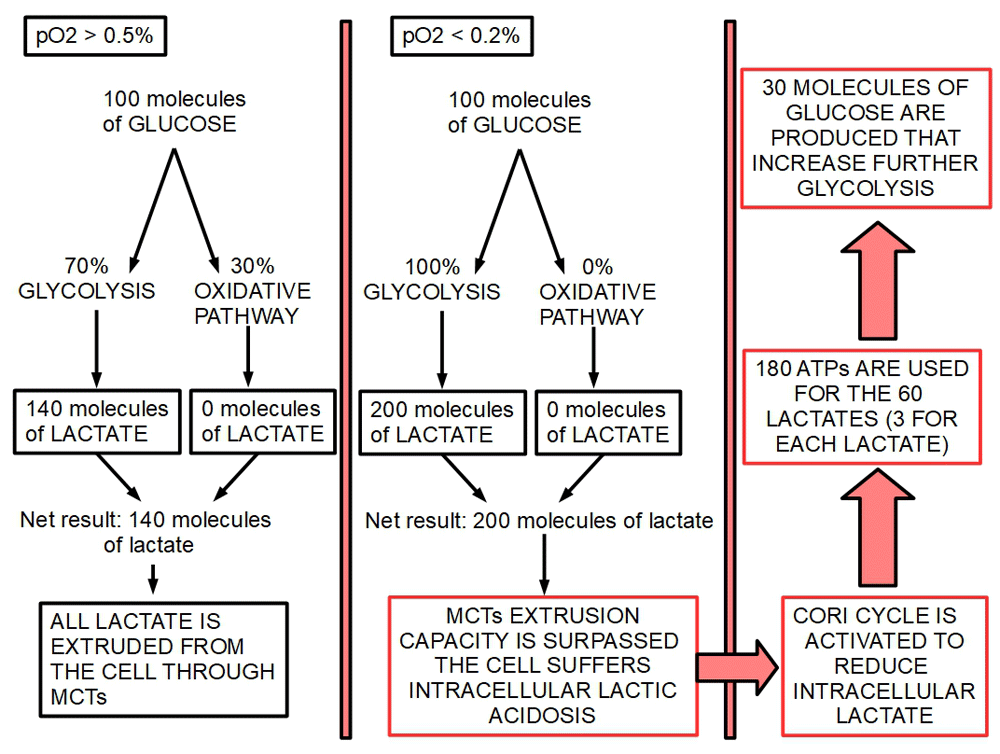
Figure 2. Energy consuming effects of the Cori cycle.
Left panel shows condition 1 (slightly hypoxic environment) while the center panel shows condition 2 (extreme hypoxia or anoxia). Right panel shows the effects of anoxia on the energy balance.
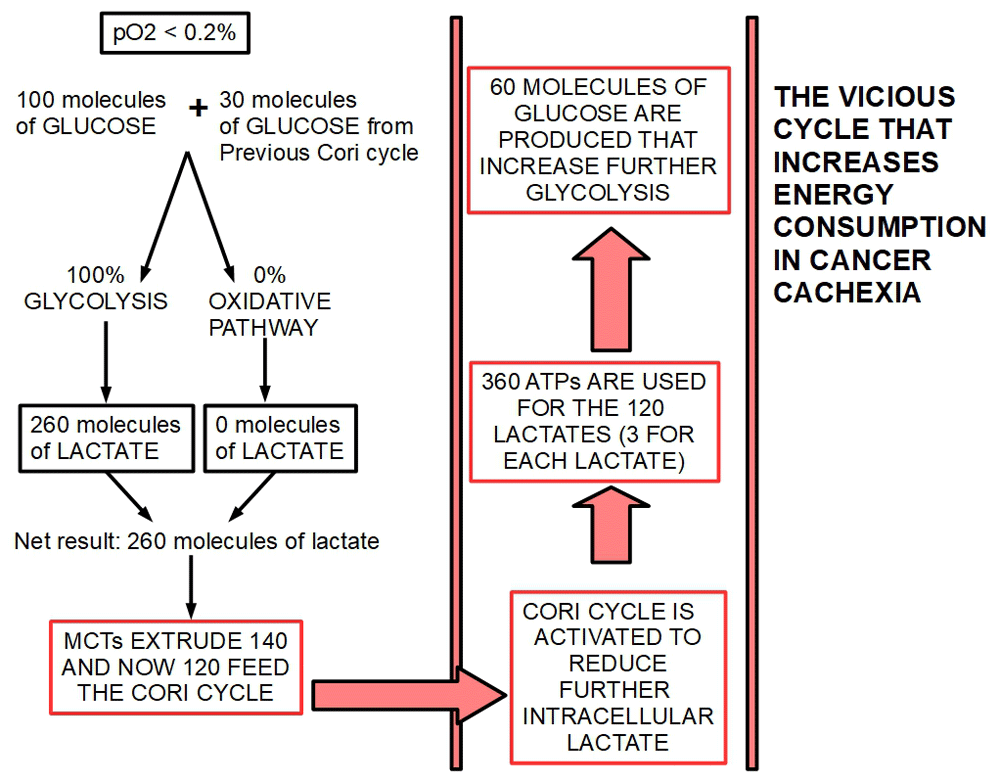
Figure 3. Energy consuming effects compounding in each Cori cycle.
The figure shows how a further turn of the cycle increases glycolytic flux and at the same time increases energy loss.
Loss of adipose tissue
Loss of adipose tissue due to increased lipolysis47 seems to be activated by protein kinase A48. Hepatic nuclear factor-4 (HNF4) mRNA was downregulated in adipose tissue of patients with CC49. Degradation of triglycerides also has a role in the loss of adipose tissue50 with the intervention of adipose triglyceride lipase51. ZAG (lipid mobilizing factor), which decreases lipids from adipocytes52, also increases the expression of uncoupling proteins in adipose tissue and skeletal muscle and therefore produce potential energy loss53,54. Interestingly, the expression of ZAG is increased with hypoxia and induces insulin resistance55.
Loss of skeletal muscle
Loss of skeletal muscle occurs due to increased proteolysis and decreased protein synthesis.
Tumor stage
Tumor stage seems to be a predictive factor of cancer cachexia56 and is probably related to tumor mass.
Insulin resistance
Asp et al.57 found that CC bearing mice had a significantly decreased glucose response to insulin. Rosiglitazone improved insulin sensitivity. Muscle wasting seems to be also related to insulin resistance58. HIF-1α can induce insulin resistance59. Intermittent hypoxia has the same effect60–63.
Tumor bioenergetics
If we look at tumor bioenergetics as a highly dynamic process that constantly adapts metabolism to oxygen and nutrients availability, we understand that most cancers have three types of cells according to their glucose metabolic behavior:
a) Oxidative when oxygen availability is high (normoxic behavior).
b) A variable mix of glycolytic and oxidative with or without hypoxia. The Warburg effect in this case is the preference for glycolytic rather than oxidative pathway.
c) Fully glycolytic when oxygen is absent (anoxic behavior).
These three types of metabolic behavior may be present in the same tumor and vary in proportion as the tumor progresses. In very advanced tumors or very bulky ones, anoxic behavior predominates and causes CC.
Figure 3 and Figure 4 represent a theoretical exercise of what would happen with 100 molecules of glucose in two different environmental conditions:
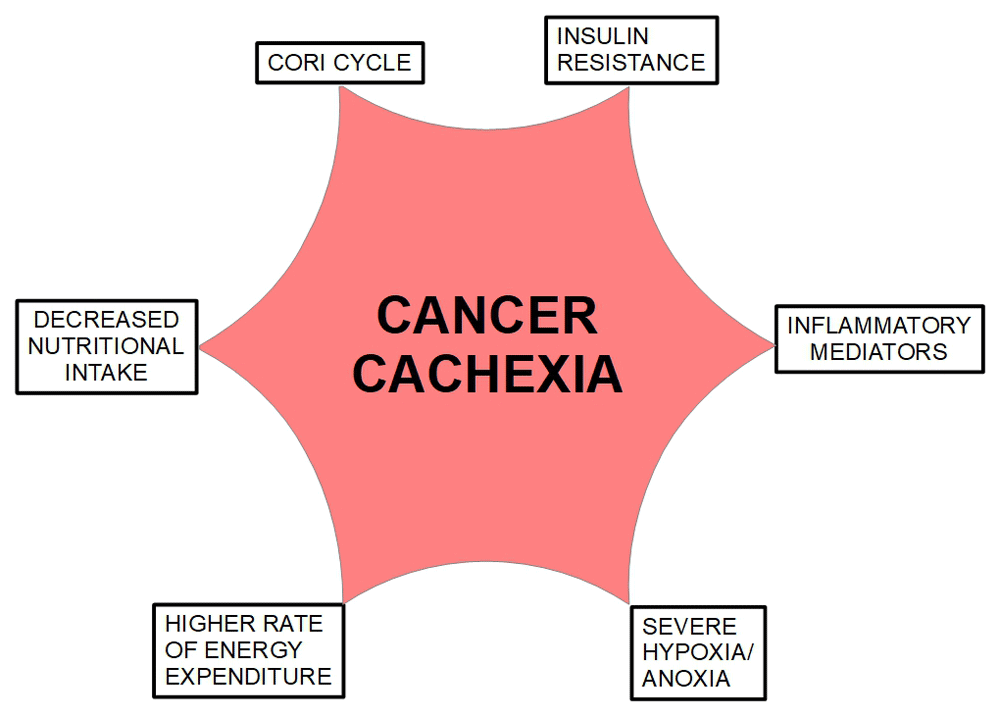
Figure 4. The causes of cancer cachexia.
These causes are showed separately. However, there is a close interaction among them. The only factor that interacts causatively with all of them is anoxia. Anoxia induces the expression and secretion of inflammatory mediators in macrophages. Anoxia also induces insulin resistance and through HIF-1α activates the Cori cycle. This cycle produces higher energy expenditure. Inflammatory mediators induce decreased nutritional intake through loss of appetite and induce skeletal muscle degradation, lipid deposit loss and finally loss of weight. All the roads lead to anoxia as the coordinator of this syndrome.
Condition 1
This condition is characterized by a pO2 higher than 0.5%. In this situation the oxidative phosphorylation would remain active and 30% or more glucose would be metabolized by mitochondria (Krebs cycle and electron transport chain) to CO2 and H2O. The rest, 70% of glucose would undergo glycolysis to lactic acid. (Warburg effect: preferential glycolytic metabolism in aerobiosis). The 140 molecules of lactate thus formed are expelled to the extracellular space by the monocarboxylate transporters. Oxidative phosphorylation remains operative during the Warburg effect. In a famous debate with Warburg64 in 1956, Sidney Weinhouse stated that Warburg’s concept about tumor cells being unable to oxidize glucose was wrong. Furthermore, Weinhouse showed that glucose can be oxidized to CO2 in cancer at a rate similar to normal cells65. This concept has since been validated by many authors66–73. The amount of oxidative phosphorylation that continues working after the metabolic shift varies considerably among different tumors and duration of hypoxia74.
What must be kept in mind is that the Warburg effect is not the shutdown of the oxidative metabolism. It is the predominance of glycolytic metabolism over oxidative metabolism, but oxidative metabolism continues working. Oxidative metabolism may be decreased, equal to or greater than in normal counterparts. The presence of oxygen increases oxidative metabolism in normal and cancer cells; however, this increase is much lower in the latter. Oxidative metabolism is present even in highly glycolytic cells but operating at a lower capacity75.
As a conclusion, the Warburg effect is not about mitochondrial metabolism impairment (as Warburg thought) but about increased glucose uptake and glycolytic flux as postulated by Weinhouse. There is high metabolic variability among cancer types and also inside a tumor. This means that glycolytic cells may conserve variable degrees of mitochondrial metabolism.
Condition 2
In this condition, the pO2 has decreased below 0.2%. Mitochondrial activity is practically downregulated by such a low level of oxygen. Therefore, nearly 100% of glucose is degraded to lactic acid, generating 200 molecules of lactate. Such a high lactate load can easily surpass monocarboxylate extruding capacity and a certain amount of lactate would remain inside the cell creating intracellular lactic acidosis that would endanger cancer cell survival. Activation of the Cori cycle comes to solve this situation by reconverting part of the lactate to glucose. This creates a vicious cycle in which the more glucose is degraded, the more the Cori cycle “works”, consuming four ATPs in each turn of the cycle. Each turn of the cycle increases the glycolytic flux by the generation of more glucose. This creates a vicious cycle. Figure 2 and Figure 3.
Figure 2 and Figure 3 are not based on real calculations. They are only used to illustrate reasons why the Cori cycle is activated and how this cycle consumes progressively higher amounts of energy. As we can see the main culprit in this scheme is extreme hypoxia.
The following concepts were taken into consideration in the construction of Figure 2 and Figure 3.
Hypoxia is an activator of gluconeogenesis (Cori cycle)
Hypobaric hypoxia has been shown to produce weight loss through diverse mechanisms including loss of appetite and activation of the Cori cycle76. HIF-1α and HIF-2α are strongly increased with ascent above 4,000 meters of altitude77. HIF-1α is a transcriptional activator of phosphoenolpyruvate carboxykinase (PEPCK) which is the rate-limiting enzyme for gluconeogenesis78. Experimental downregulation of HIF-2α decreased gluconeogenesis in hepatoma cells (HepG2) and decreased tumor size79.
The Cori cycle is a defense mechanism against lactic acidosis
Suhara et al.80 have shown that gluconeogenesis (Cori cycle) is a mechanism that defends against lactic acidosis. Why does the cancer cell need the Cori cycle? The need stems from the fact that the monocarboxylate transporter (MCT) system is saturable. The complete or almost complete abrogation of the mitochondrial metabolism plus the increased glycolytic flux represent such a burden that the MCT capacity is surpassed. In muscle, the half-maximal rate of lactate transport is achieved with a lactate concentration between 13 and 40 mM81. If the maximal rate is achieved (about 20 nmol/min per μl of intracellular volume at 25°C)82, the excess would remain inside the cell. Therefore, by transforming lactate into glucose or pyruvate The Cori cycle prevents intracellular lactic acidosis which would induce acidic stress and kill the cell. The velocity of lactate extrusion by MCTs is also dependent on intracellular and extracellular pH60. Decreased intracellular pH increases extrusion velocity, while it is lowered by extracellular acidity59. Anoxic areas of tumors have a very acidic extracellular substance, and this may slow down lactate extrusion.
The main source of glucose formed by gluconeogenesis is lactate
Koloyianni et al.83 and Ludholm et al.38 found that in normal cells, 60% of glucose generated by gluconeogenesis used lactate as the source molecule. Glutamine and alanine contributed 10% each and glycerol 3%. The rest came from serine, glycine, and threonine.
Complex I inhibitors also inhibit gluconeogenesis
Phenformin inhibits gluconeogenesis84. This is a paradoxical result in the scheme, because Complex I inhibition decreases oxidative phosphorylation. However, this anti-gluconeogenesis activity of Complex I has been tested in cells that were still performing oxidative phosphorylation. It is possible that under full anaerobiosis Complex I inhibition would have no effect on gluconeogenesis.
A proof of this last concept is that pharmacological inhibition of HIF-1α reduces cancer cachexia85. The authors used emodin and rhein (from Rheum palmatum) to decrease HIF-1α expression. Interestingly, emodin and rhein have been shown many other anti-cancer effects86–89.
Mitochondrial oxidative defect produces lactic acidemia
Mitochondrial diseases that decrease mitochondrial activity can produce lactic acidemia90. The same happens with excessive anaerobic exercise91,92. However, in the case of exercise, even though the ability of MCTs to expel lactate is not exceeded, there is no intracellular lactic acidosis.
Can anoxia by itself explain the production of inflammatory and catabolic mediators?
In 1991, Ghezzi et al.93 showed that anoxia/hypoxia with very low levels of endotoxin was able to increase levels of TNFα, IL-1α, and IL-1β more than twofold. West et al.94 further confirmed these findings and added IL-6 and prostaglandin E2 to the previous list of increased cytokine production by macrophages. Macrophages were activated in an anoxic environment95. IL-8 production is also increased in macrophages under hypoxic conditions96. All these findings were confirmed by many authors97–102. Macrophages resistant to hypoxia modify their phenotype and achieve a high production of inflammatory mediators103. It is highly possible that the inflammatory mediators found in CC are the products of macrophages associated with the tumors that are subjected to the same extreme hypoxic conditions as tumors.
Can anoxia by itself explain insulin resistance?
Insulin resistance is frequently found in patients with CC104–107. Yoshikawa et al.107 found that insulin resistance in cancer patients was not caused by malnutrition.
Intermittent hypoxia induces insulin resistance108,109. Under normal conditions insulin is a down-regulator of gluconeogenesis. With the development of insulin resistance this, inhibition is handicapped.
Usually, tumors suffer intermittent hypoxia/anoxia rather than a permanent condition. Growth, invasion and angiogenesis create a very dynamic environment with variable conditions of oxygenation110–112. Furthermore, insulin resistance is a necessary development for Cori cycle activation113, because insulin is the main downregulator of gluconeogenesis114. The following circuit is probably functional in CC:
Anoxia ➔ Insulin resistance ➔ Gluconeogenesis
TNFα is also an inducer of insulin resistance in adipocytes115 and in other tissues116,117. TNFα is a predictor of insulin resistance in pregnancy118. Saghizadeh et al. found that TNFα expression was fourfold higher in the muscle of individuals with insulin resistance compared with healthy normal controls119. Noguchi et al.120 found that TNFα increased expression was associated with insulin resistance in the skeletal muscles of cancer patients. Therefore, another circuit is probably operating in CC:
Anoxia ➔ TNFα ➔ Insulin resistance
IL6 and IL8 also play a role in insulin resistance121.
Can anoxia by itself explain lipolysis?
Briançon-Marjollet et al.122 found that endothelin-1 (ET-1) was overexpressed in adipose tissue with intermittent hypoxia and this protein activated lipolysis.
Anoxic growth versus hypoxic growth
Since the seminal works by Semenza123–126, it was well established that cells grown in hypoxic medium stabilize HIF-1α, which binds HIF-1β acting as a transcription factor dimer for genes known as hypoxia responsive genes. Therefore, stable HIF-1α expression is a signal of cellular hypoxia (with the exception of those cases where HIF-1α is constitutively activated like Von Hippel Lindau disease). If, instead of hypoxia, the tumor cell is grown under anoxia, something different happens:
1) in the first 3–10 days, HIF-1α is highly expressed;
2) after the 10th day, HIF-1α is not expressed any more127.
In both cases inflammatory cytokines are increased many folds compared with normoxic cells. The difference between the first ten days and after that is that the cell has become fully anaerobic. The authors stated “Thus, metabolically active HeLa cells respond to the lack of oxygen, in part, by regulating the levels of cytokines produced”. The increase in cytokine production was higher after 10 days of anoxia as compared with 3 days anoxia. This research clearly shows the difference in cytokines expression between short and prolonged anoxia. Figure 4 summarizes the concepts discussed above.
Usual treatments of CC
Many treatments have been used for CC. Not one has shown really encouraging results. Most of them address improving appetite and increasing food intake and/or supplementing calories. Other treatments target the intermediary chemokines such as tumor necrosis factor. Why all these failures? All the treatments used so far counter the symptoms and collateral effects of CC. None of them target the main (and unique) cause which is anoxia, or the main mechanism by which anoxia produces its effects, namely gluconeogenesis (Cori cycle). The failed therapies include:
Hydrazine sulfate, which has been tested in cachexia treatment but has not yielded any appreciable results128,129. However, some favorable results have been reported130. “Hydrazine sulfate has shown no anticancer activity in randomized clinical trials, and data concerning its effectiveness in treating cancer-related cachexia are inconclusive”131. It has not been approved by the FDA for any medical condition.
Steroids. Different steroids such as medroxyprogesterone132–134, megestrol135–137 have been tested for cachexia treatment. Megestrol have shown beneficial effects limited to appetite, however it does not impact cachexia and is associated with many side effects138.
Eicosapentaenoic acid. This is an omega-3 (n-3) polyunsaturated fatty acid (PUFAs). It targets the loss of muscular mass, but does not solve the other effects of CC139,140.
High calorie nutritional supplements.
High dose progestins. These did lead to some appetite and weight improvement but without major results in the relentless evolution of CC141.
Etanercept and infliximab have been used as anti-TNFα with poor results in CC142.
Tocilizumab is an anti-IL6 antibody approved by the FDA for rheumatoid arthritis treatment. It showed benefits in some cases of CC143–145. However, there are no randomized clinical studies confirming these benefits. The number of cases that have been published are scarce.
Insulin for the treatment of insulin resistance. Lundholm et al.146 found insulin as an important palliative treatment for patients with cancer-related weight loss.
Rosiglitazone for insulin resistance treatment.
COX2 inhibitors like celecoxib for decreasing acute phase pro-inflammatory cytokines. A pilot study with celecoxib showed beneficial effects in patients with CC147.
In the next section we shall propose a treatment scheme based on targeting the physiopathology of CC, rather than the secondary symptoms.
Discussion and hypothesis
From a metabolic point of view, there are three types of cells in most tumors:
a) Oxidative cells located near blood vessels with adequate or near adequate oxygen and nutritional supply.
b) Aerobic glycolytic cells located in the tumor mass with inadequate oxygen supply but with a functional oxidative phosphorylation that metabolizes part of the glucose to CO2 and H2O; however, these cells preferentially and in major proportion use the glycolytic pathway to lactic acid.
c) Deeply anaerobic cells with near zero supply of oxygen in which only glycolysis to lactic acid is functional. These cells are unable to perform oxidative phosphorylation.
The proportion of each of these phenotypes in a tumor are variable and dynamic. At an early stage and in small tumors probably oxidative and glycolytic aerobic cells are found. As the tumor continues growing, the severely anaerobic cells, appear. The reason for this is mainly anatomic: they are in completely oxygen deprived areas. Since this last group of cells is incapable of using oxidative phosphorylation it is fully glycolytic and its lactic acid output is higher than in the other two groups.
Fully anaerobic cells have three characteristics:
a) very high level of HIF-1α expression and activation;
b) high level of intracellular lactic acid which surpasses the extrusion capacity of monocarboxylate transporters;
c) a tendency towards intracellular lactic acidosis.
HIF-1α upregulates PEPCK, activating the Cori cycle. This creates an increased energy imbalance due to a loss of four ATPs for each complete glucose-lactate-glucose cycle. In spite of the energy imbalance thus created, the new scheme rescues the cell from death due to intracellular acidification and restores the intracellular alkalinity needed for adequate proliferation.
When the proportion of severely anaerobic cells in a tumor increases, cancer cachexia develops. Usually this is the result of tumors with poor vascular supply and/or large size.
Many pro-cachexia tumors produce proteins and cytokines (whether by themselves or by stimulating other tissues) that have a lipolytic or miolytic effect such as myostatin148. TNFα and interferon-γ produce loss of appetite and consequently decreased food intake149.
What is the evidence sustaining the above hypothesis?
1) The lactate shuttle is the best proof of the coexistence of oxidative cells and aerobic glycolytic cells.
2) Frequent findings of necrotic areas in large tumors prove the existence of cells that are extremely anoxic but could not implement the salvage through the Cori cycle.
3) Cancer cachexia frequently appears in the late stages of malignant progression.
4) Cancer cachexia appears progressively, as certain tumors increase in size150.
The sequence of events leading to CC is shown in Figure 5 and Figure 6.
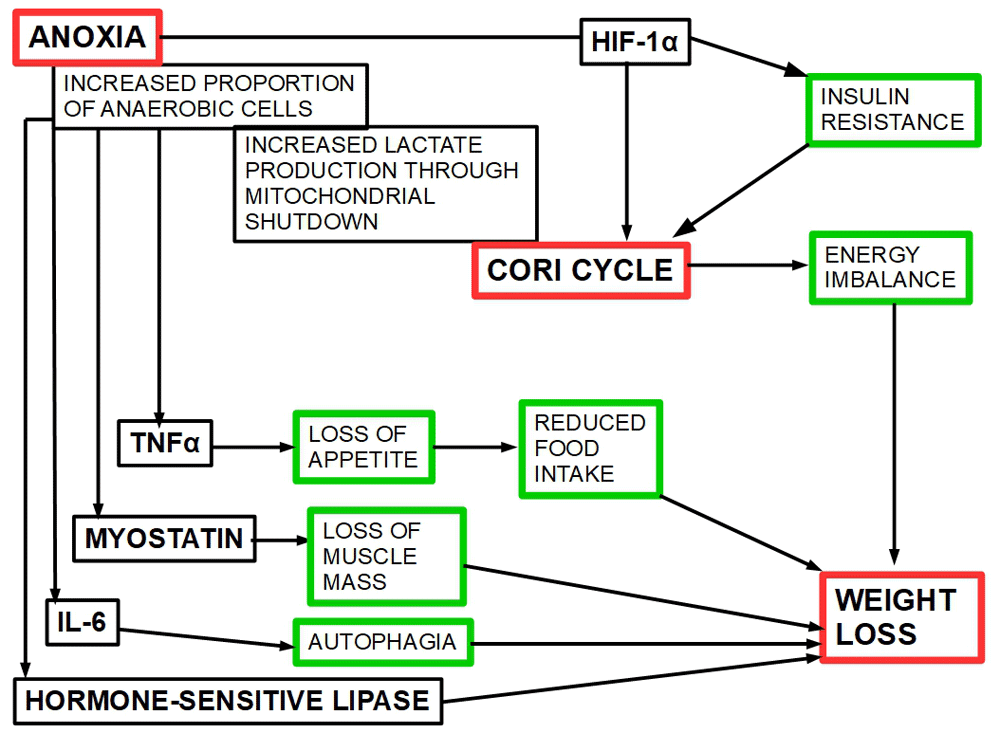
Figure 5. Sequence of events starting in anoxia and leading to weight loss.
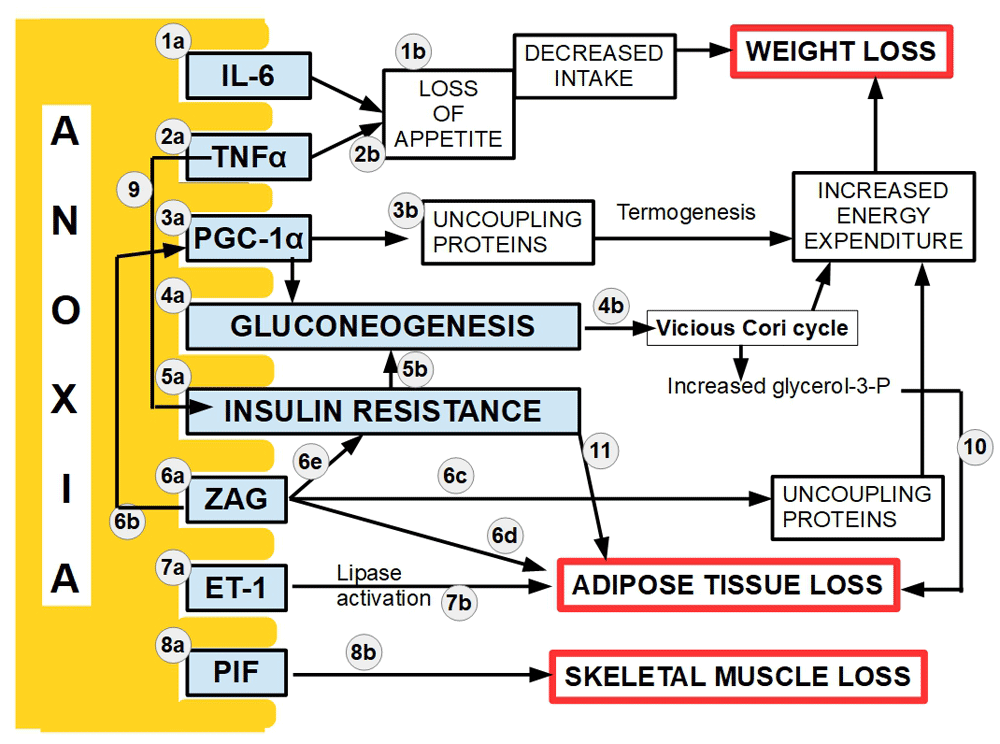
Figure 6. An integrated view of the factors intervening in CC and their relation to anoxia.
1a, 2a, 3a, 4a, 5a, 6a,7a, and 8a indicate the relation of anoxia/hypoxia on one side to the chemokines (also called toxohormones by some authors) or direct metabolic effects on the other side.
The mediator action of cytokines produced by the anoxic cells is related to loss of appetite, loss of muscle and adipose mass. HIF-1α is the transcription factor that activates the Cori cycle as a salvage mechanism from the lactate overload. The center of all these activities is extreme hypoxia/anoxia/intermittent anoxia.
Figure 6 is an integrated view of the relationship between anoxia/deep hypoxia/intermittent hypoxia, on one side and the intermediaries leading to the cardinal symptoms of CC. Also the relationship among these intermediaries has been included in the drawing. This figure shows the essential link between anoxia and CC. The figure is based on the following references: References on Figure 6: 1a)151–155, 1b)156–159; 2a)160–165; 2b)166–169; 3a)170–172; 3b)173,174; 4a)80,175–177; b)178–180; 5a)62,63,181–184; 5b)185,186; 6a)187–191; 6b)192; 6c)193–195; 6d)196–200; 6e)57; 7a)126. IL-6 stimulates the production of endothelin-1 (ET-1)201, not shown in the figure. ET-1 is also a potent vasoconstrictor increasing anoxia202, not shown in the figure; 7b)126,203,204. ET-1 also induces insulin resistance205, not shown in the figure; 8a)206; 8b)207–211; 9)13; 10) Increased glycerol-3-phosphate production, whether from glycolysis or the Cori cycle stimulate adipose tissue loss under hypoxia212; 11) Insulin resistance increases lipolysis213,214.
The role of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α)
PGC-1α is a 798-amino-acid transcriptional coactivator considered an important activator of mitochondrial biogenesis215,216. Interestingly, this protein has some characteristics that are important in CC:
PGC-1α is a coactivator for the transcription of other proteins that act in energy metabolism.
It determines lactate metabolism217.
It activates mitochondrial fatty acid β oxidation218,219.
PGC-1α induces gluconeogenesis220.
It activates thermogenic genes, increasing energy expenditure221.
PGC-1α can be recruited by estrogen related receptors222,223.
And most importantly, it is increased in hypoxic conditions224 in different tissues including central nervous system225.
PGC-1α, a hypoxia inducible coactivator protein increases thermogenesis and loss of energy and induces mitochondrial lipolysis. PGC-1α shows also other pro-tumoral activities in:
Glioblastoma, where it determines a more aggressive phenotype226.
ER-negative breast tumors, in which the level of estrogen related receptors is significantly increased and probably interacting with PGC-1α. Furthermore, the association of PGC-1α with estrogen related receptors positively regulates HIF-2α transcription227.
Colorectal cancer, in which hypoxia induced over-expression of PGC-1α regulates tumorigenesis, enhancing cell motility, proliferation, stemness, resistance to chemotherapy, and reducing ROS by antioxidant enzymes activation228.
Melanoma, which when PGC-1α is overexpressed, cells show oxidative metabolism229,230.
Our interpretation of PGC-1α over-expression in the CC is that cells try to activate the mitochondrial oxidative metabolism in a process where anoxia has near shut down this metabolic pathway. Therefore, PGC-1α is a hypoxia modulated protein that is able to control many of the intermediary steps between anoxia/hypoxia and the symptoms of CC.
Treatment proposal
In this paper we argue that anoxia is the main cause of CC. Figure 5 and Figure 6 shows how anoxia achieves all the cardinal symptoms of CC through diverse mechanisms. The figure does not show the intracellular lactic acidosis, because the Cori cycle prevents its onset.
Based on Figure 5 and Figure 6 we must first target anoxia and the Cori cycle. Then we can attack the symptoms. Going against the symptoms alone has failed consistently in the past.
The scheme proposed here:
Anoxia is difficult to target. Vasodilators such as nitrates and “hemodynamic improvers” such as pentoxifylline may improve circulation in the tumor decreasing anoxia. Flunarizine is a vasodilator with the ability to block voltage gated sodium channels. Nitroglycerin is another vasodilator that has been tested in tumors. A combination of these drugs should enhance tumor oxygenation. Any anti-angiogenic drugs being used, should be discontinued.
HIF-1α, directly upregulated by anoxia, is a targetable transcription factor. Although extensive and intensive research for an inhibitor has been going on for many years, no adequate drug has been developed yet. Emodin is a non-toxic inhibitor of HIF and may produce some benefits231–235 in CC.
Intracellular lactic acidosis-Cori cycle-gluconeogenesis: one of the mechanisms to decrease Cori cycle is to impede excessive lactic acid production, whether in the anaerobic cancer cells or in the liver. In this sense probably the drug of choice should be dichloroacetate (DCA).
Improving appetite: if the previous issues have been medicated and controlled to a certain extent, then improving appetite with megestrol and administering high calorie nutritional supplements makes sense.
The fundamentals for the treatment scheme
Metabolic modifier: DCA
DCA is an investigational drug for lactic acidosis, pulmonary hypertension and now for cancer. In some African countries, its use is unofficially accepted for the treatment of lactic acidosis in children with malaria236. It is a small molecule with the following formula HOOC-CH2-Cl2. DCA is an orally available molecule that is almost completely and quickly absorbed at the digestive system237,238. It is metabolized by GSTZ1 (a glutathione transferase isoform)239.
When DCA was administered to 16 healthy individuals, in 1 to 50 mg/kg IV infusions, it lowered plasma glucose, lactate and alanine concentrations. Plasma levels linearly followed those of the administered dose up to 30 mg/kg. A dose of 35 mg/kg was considered most effective regarding lactic acid, which fell to 75% below baseline concentrations within 2 hours of the infusion240. Blood glucose was not affected in these healthy individuals but was reduced in diabetic patients through stimulation of peripheral glucose utilization and inhibition of gluconeogenesis. DCA also inhibits lipogenesis and cholesterol synthesis. Daily oral administration of 50 mg/kg DCA to diabetic patients with slightly elevated lactic acid concentration, reduced alanine and lactic acid in plasma241.
The experimental administration of a single dose of DCA may be misleading upon dose and metabolism, because as Gonzalez-Leon et al.242 have shown in rats with repeated administration of DCA, this drug has a surprising feature: it inhibits its own metabolism. This means that the chronic administration of DCA differs from single doses in its plasma concentration, toxicology and metabolism. Figure 7.
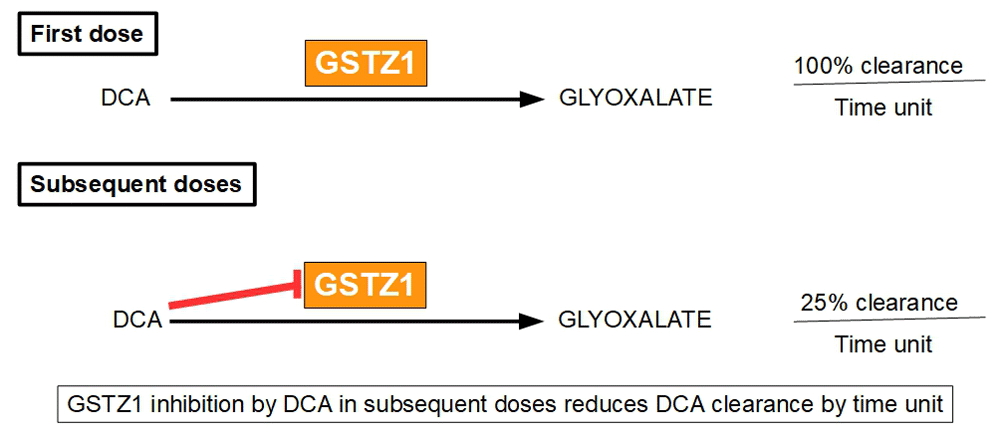
Figure 7. Differences in DCA clearance between first and subsequent administration.
This is an important issue to be considered for the clinical use of DCA.
In humans, the maximum plasma concentration achieved with an IV infusion of 10 mg/kg was between 19.9 μg/ml and 24.7 μg/ml with a half life of only 20 minutes. When the infused dose was increased to 20 mg/kg the plasma concentration was between 57.3 and 74.9 μg/ml with a half life of 36 minutes. In dogs and rats the half life was much longer (the half life of a 100 mg/kg dose was 4 hours and 24 hours in dogs and rats respectively243. These marked differences among species cast doubts about the possibility of translating the findings in other mammals to humans.
Confirming these inter-species differences, Maissenbacher et al. have found that dogs present enhanced inhibition of DCA degradation and slower clearance than humans and rats due to increased inhibition of GSTZ1244. The DCA metabolic pathway starts with dehalogenation to monochloroacetate and glyoxylate and then it continues to glycine, and the final products are oxalate and carbon dioxide245. The first dose is cleared from plasma faster than subsequent doses, as Gonzalez-Leon et al. have shown; this is probably due to GSTZ1 inhibition by DCA in subsequent doses. This is similar in all species. The decrease in DCA clearance by multiple doses is not a minor issue, because the initial clearance may be reduced to less than 25% of the initial one in successive doses246.
The main mechanism of action of DCA is the inhibition of pyruvate dehydrogenase kinase (PDK) and its isoforms. This inhibition increases the flux of pyruvate into the mitochondria, promoting glucose oxidation instead of glycolysis247. PDK inactivates the pyruvate dehydrogenase enzyme complex through phosphorylation. By downregulating the activity of this complex, PDK decreases the oxidation of pyruvate in mitochondria and increases the conversion of pyruvate to lactate in the cytosol.
Other mechanisms of action of DCA in cancer can also be found in the medical literature. Stockwin et al.248 described that cytotoxicity of DCA is only achieved in those cells that suffered mitochondrial DNA mutations that “condemn” them exclusively to the glycolytic pathway. Therefore, DCA has features that make presume it will reduce glycolysis, lactic acid production and gluconeogenesis in anaerobic malignant cells or even cause their death.
Why DCA?
TNFα and IL-1α are inhibitors of pyruvate dehydrogenase and mitochondrial metabolism249,250. DCA has exactly the contrarian effect.
DCA decreases the expression of TNFα and IL-1β and lactate production in ischemic insults251. It also decreases the expression of IL-6 and Interferon γ252.
While TNFα increases fermentative glycolysis, DCA decreases it253.
DCA is an inhibitor of lipolysis254.
DCA decreases all the gluconeogenic precursor molecules255 and thus probably decreasing gluconeogenesis.
DCA seems to reduce insulin resistance256.
It has been suggested that DCA had the ability to decrease/block the Cori cycle257,258.
DCA showed inhibitory effects on HIF-1α in glioblastoma cells259,260.
DCA synergizes with other chemotherapeutic drugs261–266.
DCA targets mainly cells that cannot use the oxidative metabolism248. This concept is further confirmed by the synergy between DCA and metformin267–274. The fundamentals of this association stem from the fact that metformin inhibits mitochondrial Complex I and reduces oxidative metabolism while DCA inhibits glycolysis. This double-edged approach would target very hypoxic cells where oxidative metabolism is minimal and is even further blocked by metformin.
According to the effects discussed above, DCA seems the only drug that targets simultaneously most of the proteins and pathways involved in CC pathogenesis.
Increasing tumor oxygenation and inhibiting TNFα: Pentoxifylline, nitroglycerin and thalidomide
Pentoxifylline reduces the expression of TNFα in cancer cells. It has been tested in CC in five patients with good results in three of them275. Many other reports have confirmed the inhibitory actions of pentoxifylline on TNFα276–279, not only in cancer, but also in other pathologies280,281. Pentoxifylline has other very important actions in CC: it is a hemorheological agent that increases red blood cell deformability, reduces blood viscosity and decreases platelet aggregation improving microcirculation282. Therefore, a better oxygenation of the tumor is expectable. However, a study by Goldberg et al.283 with pentoxifylline as a stand-alone drug did not show any improvement in patients with CC. There is evidence that pentoxifylline increases tumor oxygenation284,285.
Nitroglycerin (NTG) is another drug that deserves consideration. It is a well-known vasodilator and oxygenation improver. In addition to these effects NTG also reduces HIF-1α levels in hypoxic tumors286,287, because it acts as a nitrous oxide donor. Unfortunately, NTG has both pro and anti-tumor effects288. However, used on a short-term basis as an adjunct to pentoxifylline, an important improvement of tumor oxygenation can be expected. For a review of nitroglycerin’s anti-tumoral activity, read Sukhatme et al.289.
Thalidomide is a TNFα downregulator290 and has been tested in CC with encouraging perspectives291. It had similar results in wasting syndromes of other origins292,293. Thalidomide has also other anti-cancer effects294, such as anti-angiogenesis and T-cell stimulation295, which will not be considered here because they go beyond the scope of this manuscript. It has been established as part of multiple myeloma treatment protocols. For a review of thalidomide, read Luzzio296.
Downregulation/inhibition of HIF-1α
As mentioned above, emodin can be used for this purpose. Emodin down-regulates HIF-1α expression297. It also inhibits pro-inflammatory responses298 that play a role in CC. Furthermore, hepatic cancer cells (HepG2) treated with emodin showed a significant decrease of lactate. This decrease is a signal of glycolytic inhibition which has been further confirmed because emodin decreased mRNA levels of hexokinase II (HKII), pyruvate kinase isoform M2 (PKM2), and lactate dehydrogenase-A (LDHA) in a concentration-dependent manner299. Emodin also inhibits TNFα, NF-κB and IL-6, all mediators of CC300. Emodin has many other anti-cancer effects, such as:
sensitization to chemotherapeutics235,301–303;
increasing ROS production304;
promoting apoptosis;
inhibiting angiogenesis305,306, metastasis307, migration and invasion308:
inducing proteasomal degradation of Her2/neu309;
inhibiting ATP citrate lyase310;
increasing expression of insulin-like growth factor binding protein-1311;
reverting cisplatin resistance312;
blocking STAT 3 activation313, among others.
For a review on emodin pharmacology see Dong et al.314.
The treatment proposed here includes the association of pentoxifylline, emodin and DCA as an added scheme to classical treatments such as high calorie nutritional supplements and anabolics like megestrol (Figure 8). Adding these drugs to the conventional treatment would decrease anoxia, HIF-1α expression, and the glycolytic pathway restoring oxidative phosphorylation and reducing TNFα expression. This type of treatments targets the etiology of CC rather than the symptoms.
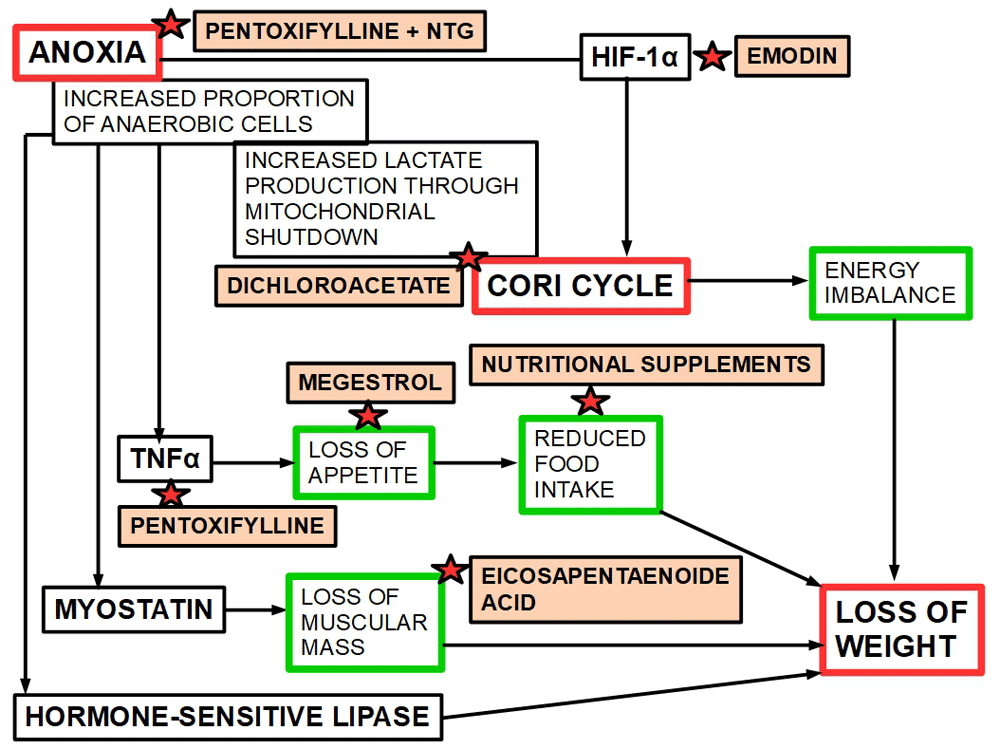
Figure 8. A diagram showing the specific site of action of each drug with anti-CC potential.
Inhibition of PEPCK to block gluconeogenesis
PEPCK is the rate-limiting enzyme for gluconeogenesis. Therefore, its inhibition should block the Cori cycle. Many drugs have been identified with the ability to inhibit PEPCK, such as metformin315, troglitazone316, berberine317, among others. None of these drugs have been tested in CC. Berberine should be considered a particularly interesting drug because it inhibits PEPCK but also downregulates HIF-1α318. Berberine also has many other anti-cancer effects319, such as down-regulation of COX2320, increased apoptosis in cancer cells without affecting the normal ones321,322, reduced migration and invasion, among others. For a review of other anti-cancer effects of berberine, read Kaboli et al.323
Other potential drugs
3-bromopyruvate (3BP) is a protein alkylating agent that has shown many anti-cancer effects. We included it in this list of possible drugs for treating CC because it is a potent inhibitor of aerobic glycolysis324. Inhibition of glycolysis should decrease lactate production, thus decreasing the Cori cycle. The exact mechanism of action is not fully known, but there is some evidence pointing to inhibition of glycolytic enzymes325. It has important cytotoxic effects on highly glycolytic tumor cells326.
Tocilizumab (a humanized anti-IL6 receptor antibody)327 may produce some benefits328,329. It requires further testing. The association of tocilizumab with gemcitabine for the treatment of advanced pancreatic cancer failed to show clear clinical benefit in a phaseI/II clinical Trial330.
Insulin, besides its known actions (inhibitor of gluconeogenesis), exerts inhibitory activity on PGC1α expression331.
Anamorelin is a small molecule ghrelin receptor agonist that has shown favorable effects on appetite, food intake and weight gain in patients with CC332–334. However, its approval was rejected twice by the European Medicines Agency335.
An alternative hypothesis: the browning of adipose tissue
Petruzzelli et al.336 reported a completely different physiopathological road leading from cancer to cachexia: the browning of white adipose tissue. They maintained that a phenotypic switch from white adipose tissue to brown adipose tissue metabolism was the main culprit of CC. The main characteristic of browning would be the increased expression of uncoupling protein-1 in white adipose tissue, with consequent high energy expenditure. They also found that inflammatory intermediaries (mainly IL-6) were the cause of the browning process and proposed the anti-inflammatory sulindac for CC treatment. Considering that the findings of Petruzzelli et al. are correct, one question remains unanswered: why do advanced tumors produce significant inflammatory mediators? And this takes as back to the anoxia problem: it is anoxia that induces the production of inflammatory mediators.
The steps between anoxia and CC can be those proposed by Petruzzelli et al. (browning of white adipose tissue) or those hypothesized in this paper (increased Cori cycle). The increased Cori cycle can occur in the tumor, in the liver or in both. However, the primum movens remains anoxia. Treating the intermediate steps (TNFα, IL-6 or other chemokines), or the symptoms (loss of weight, lipolysis, muscle loss) are valid approaches. However, the only significant result would be achieved by simultaneously targeting fermentative glycolysis and anoxia alongside to the other treatments.
A unified explanation of the causes of CC has not been achieved yet337. Therefore, anoxia as the unifying cause behind CC deserves more research.
Conclusions
A unitary explanation of the cause of CC is presented here. The main culprit of this wasting syndrome is anoxia. The molecular mechanisms leading from anoxia to the full blown syndrome are also presented. A therapeutic approach, based on this hypothesis is proposed.
Anoxia in large areas of the tumor mass is the main cause of CC. This occurs through a sequence of events where oxidative phosphorylation is almost totally shut down leading to full glycolytic behavior (100% of the glucose is degraded through fermentation and none through oxidation). Vascular supply and cell metabolism are highly heterogeneous throughout the tumor. Anoxic anaerobic metabolism is also present in parts of the tumoral mass. When an important portion of the tumor is “pushed” to fully anaerobic metabolism by lack of oxygen, CC develops. Even the most recent publications on CC miss the central issue: anoxia. Therefore, there is no place for anti-anoxic treatments in the therapeutic protocols being used routinely. Anoxia produces such a high level of intracellular lactate that it surpasses monocarboxylate transporters extruder capacities. Thus, the Cori cycle is triggered to prevent intracellular lactic acidosis creating an energetic imbalance due to the cycle’s high energy requirements. Increased inflammatory mediators, that cause many of the symptoms of CC, are not produced by the tumor itself but by hypoxia resistant macrophages associated to the malignant stroma.
All the treatments employed up to now have failed because they addressed the symptoms of CC instead of the causes. Here we propose targeting anoxia, HIF-1α, and the glycolytic pathway as the logical treatment for CC using a combination of drugs such as pentoxifylline, dichloroacetate, metformin and emodin associated with anabolic steroids and nutritional supplements. The drugs can be changed for others with a similar effect. What is important is to center the treatment on tumoral anoxia, the glycolytic pathway and TNF. It is probably useless to address only one of these issues or expect real improvements with nutritional supplements and appetite improvers. The usually late onset of CC in a prolonged disease and the frequent therapeutic failures have paved the way for a nihilistic attitude that has prevailed up to the present. Targeting the strongly anaerobic cells in the tumor will not only improve CC but at the same time slow down the disease and eventually prolong survival. DCA and the association of DCA with metformin, vasodilators, and HIF-1α inhibitors deserve well planed experimental and clinical research for CC’s therapy.
Data availability
No data are associated with this article.
Faculty Opinions recommendedReferences
- 1.
Bennani-Baiti N, Walsh D:
What is cancer anorexia-cachexia syndrome? A historical perspective.
J R Coll Physicians Edinb.
2009; 39(3): 257–262. PubMed Abstract
- 2.
Tisdale MJ:
Cachexia in cancer patients.
Nat Rev Cancer.
2002; 2(11): 862–71. PubMed Abstract
| Publisher Full Text
- 3.
Fearon K, Arends J, Baracos V:
Understanding the mechanisms and treatment options in cancer cachexia.
Nat Rev Clin Oncol.
2013; 10(2): 90–9. PubMed Abstract
| Publisher Full Text
- 4.
Fearon K, Strasser F, Anker SD, et al.:
Definition and classification of cancer cachexia: an international consensus.
Lancet Oncol.
2011; 12(5): 489–495. PubMed Abstract
| Publisher Full Text
- 5.
Evans WJ, Morley JE, Argilés J, et al.:
Cachexia: a new definition.
Clin Nutr.
2008; 27(6): 793–799. PubMed Abstract
| Publisher Full Text
- 6.
Sarhill N, Mahmoud F, Walsh D, et al.:
Evaluation of nutritional status in advanced metastatic cancer.
Support Care Cancer.
2003; 11(10): 652–659. PubMed Abstract
| Publisher Full Text
- 7.
Bruera E, Sweeney C:
Cachexia and asthenia in cancer patients.
Lancet Oncol.
2000; 1(3): 138–147. PubMed Abstract
| Publisher Full Text
- 8.
Theologides A:
Cancer cachexia.
Cancer.
1979; 43(5 Suppl): 2004–2012. PubMed Abstract
| Publisher Full Text
- 9.
Olson SH, Xu Y, Herzog K, et al.:
Weight Loss, Diabetes, Fatigue, and Depression Preceding Pancreatic Cancer.
Pancreas.
2016; 45(7): 986–91. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 10.
Wu YS, Lin PY, Chien CY, et al.:
Anxiety and depression in patients with head and neck cancer: 6-month follow-up study.
Neuropsychiatr Dis Treat.
2016; 12: 1029–36. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 11.
Langius JA, Twisk J, Kampman M, et al.:
Prediction model to predict critical weight loss in patients with head and neck cancer during (chemo)radiotherapy.
Oral Oncol.
2016; 52: 91–96. PubMed Abstract
| Publisher Full Text
- 12.
Barreto R, Mandili G, Witzmann FA, et al.:
Cancer and Chemotherapy Contribute to Muscle Loss by Activating Common Signaling Pathways.
Front Physiol.
2016; 7: 472. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 13.
Patel HJ, Patel BM:
TNF-α and cancer cachexia: Molecular insights and clinical implications.
Life Sci.
2017; 170: 56–63. PubMed Abstract
| Publisher Full Text
- 14.
Lowry SF, Moldawer LL:
Tumor necrosis factor and other cytokines in the pathogenesis of cancer cachexia.
PrincPract Oncol.
1990; 4: 1–12.
- 15.
Oliff A, Defeo-Jones D, Boyer M, et al.:
Tumors secreting human TNF/cachectin induce cachexia in mice.
Cell.
1987; 50(4): 555–563. PubMed Abstract
| Publisher Full Text
- 16.
Ebadi M, Mazurak VC:
Potential Biomarkers of Fat Loss as a Feature of Cancer Cachexia.
Mediators Inflamm.
2015; 2015: 820934. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 17.
Mondello P, Mian M, Aloisi C, et al.:
Cancer cachexia syndrome: pathogenesis, diagnosis, and new therapeutic options.
Nutr Cancer.
2015; 67(1): 12–26. PubMed Abstract
| Publisher Full Text
- 18.
Uehara A, Sekiya C, Takasugi Y, et al.:
Anorexia induced by interleukin 1: involvement of corticotropin-releasing factor.
Am J Physiol.
1989; 257(3 Pt 2): R613–R617. PubMed Abstract
| Publisher Full Text
- 19.
Blumberg D, Hochwald S, Brennan MF, et al.:
Interleukin-6 stimulates gluconeogenesis in primary cultures of rat hepatocytes.
Metabolism.
1995; 44(2): 145–146. PubMed Abstract
| Publisher Full Text
- 20.
Martínez-Hernández PL, Hernanz-Macías Á, Gómez-Candela C, et al.:
Serum interleukin-15 levels in cancer patients with cachexia.
Oncol Rep.
2012; 28(4): 1443–1452. PubMed Abstract
| Publisher Full Text
- 21.
Anandavadivelan P, Lagergren P:
Cachexia in patients with oesophageal cancer.
Nat Rev Clin Oncol.
2016; 13(3): 185–98. PubMed Abstract
| Publisher Full Text
- 22.
Loumaye A, de Barsy M, Nachit M, et al.:
Role of Activin A and myostatin in human cancer cachexia.
J Clin Endocrinol Metab.
2015; 100(5): 2030–2038. PubMed Abstract
| Publisher Full Text
- 23.
Greco SH, Tomkötter L, Vahle AK, et al.:
TGF-β Blockade Reduces Mortality and Metabolic Changes in a Validated Murine Model of Pancreatic Cancer Cachexia.
PLoS One.
2015; 10(7): e0132786. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 24.
Banks WA:
Anorectic effects of circulating cytokines: role of the vascular blood-brain barrier.
Nutrition.
2001; 17(5): 434–437. PubMed Abstract
| Publisher Full Text
- 25.
Iwase S, Murakami T, Saito Y, et al.:
Steep elevation of blood interleukin-6 (IL-6) associated only with late stages of cachexia in cancer patients.
Eur Cytokine Netw.
2004; 15(4): 312–316. PubMed Abstract
- 26.
Pettersen K, Andersen S, Degen S, et al.:
Cancer cachexia associates with a systemic autophagy-inducing activity mimicked by cancer cell-derived IL-6 trans-signaling.
Sci Rep.
2017; 7(1): 2046. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 27.
Bosaeus I, Daneryd P, Svanberg E, et al.:
Dietary intake and resting energy expenditure in relation to weight loss in unselected cancer patients.
Int J Cancer.
2001; 93(3): 380–383. PubMed Abstract
| Publisher Full Text
- 28.
Nguyen TY, Batterham MJ, Edwards C:
Comparison of Resting Energy Expenditure Between Cancer Subjects and Healthy Controls: A Meta-Analysis.
Nutr Cancer.
2016; 68(3): 374–387. PubMed Abstract
| Publisher Full Text
- 29.
Dev R, Hui D, Chisholm G, et al.:
Hypermetabolism and symptom burden in advanced cancer patients evaluated in a cachexia clinic.
J Cachexia Sarcopenia Muscle.
2015; 6(1): 95–98. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 30.
Ryan AM, Power DG, Daly L, et al.:
Cancer-associated malnutrition, cachexia and sarcopenia: the skeleton in the hospital closet 40 years later.
Proc Nutr Soc.
2016; 75(2): 199–211. PubMed Abstract
| Publisher Full Text
- 31.
Penet MF, Bhujwalla ZM:
Cancer cachexia, recent advances, and future directions.
Cancer J.
2015; 21(2): 117–22. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 32.
Porporato PE:
Understanding cachexia as a cancer metabolism syndrome.
Oncogenesis.
2016; 5(2): e200–e200. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 33.
Friesen DE, Baracos VE, Tuszynski JA:
Modeling the energetic cost of cancer as a result of altered energy metabolism: implications for cachexia.
Theor Biol Med Model.
2015; 12(1): 17. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 34.
Nelson DL, Cox MM:
Lehninger Principles of Biochemistry Fourth Edition. New York: W.H. Freeman and Company. 2005. Reference Source
- 35.
Waterhouse C:
Lactate metabolism in patients with cancer.
Cancer.
1974; 33(1): 66–71. PubMed Abstract
| Publisher Full Text
- 36.
Holroyde CP, Skutches CL, Boden G, et al.:
Glucose metabolism in cachectic patients with colorectal cancer.
Cancer Res.
1984; 44(12 Pt 1): 5910–5913. PubMed Abstract
- 37.
DeWys WD:
Pathophysiology of cancer cachexia: current understanding and areas for future research.
Cancer Res.
1982; 42(2 Suppl): 721s–726s. PubMed Abstract
- 38.
Lundholm K, Edström S, Karlberg I, et al.:
Glucose turnover, gluconeogenesis from glycerol, and estimation of net glucose cycling in cancer patients.
Cancer.
1982; 50(6): 1142–1150. PubMed Abstract
| Publisher Full Text
- 39.
Gadducci A, Cosio S, Fanucchi A, et al.:
Malnutrition and cachexia in ovarian cancer patients: pathophysiology and management.
Anticancer Res.
2001; 21(4B): 2941–2947. PubMed Abstract
- 40.
Tisdale MJ:
Cancer cachexia: metabolic alterations and clinical manifestations.
Nutrition.
1997; 13(1): 1–7. PubMed Abstract
| Publisher Full Text
- 41.
Tayek JA, Katz J:
Glucose production, recycling, Cori cycle, and gluconeogenesis in humans: relationship to serum cortisol.
Am J Physiol.
1997; 272(3 Pt 1): E476–E484. PubMed Abstract
| Publisher Full Text
- 42.
Passarella S, Schurr A:
l-Lactate Transport and Metabolism in Mitochondria of Hep G2 Cells-The Cori Cycle Revisited.
Front Oncol.
2018; 8: 120. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 43.
Vazeille C, Jouinot A, Durand JP, et al.:
Relation between hypermetabolism, cachexia, and survival in cancer patients: a prospective study in 390 cancer patients before initiation of anticancer therapy.
Am J Clin Nutr.
2017; 105(5): 1139–1147. PubMed Abstract
| Publisher Full Text
- 44.
Katz J, Tayek JA:
Gluconeogenesis and the Cori cycle in 12-, 20-, and 40-h-fasted humans.
Am J Physiol.
1998; 275(3): E537–E542. PubMed Abstract
| Publisher Full Text
- 45.
Hall KD, Baracos VE:
Computational modeling of cancer cachexia.
Curr Opin Clin Nutr Metab Care.
2008; 11(3): 214. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 46.
Bongaerts GP, Van Halteren HK, Verhagen CA, et al.:
Cancer cachexia demonstrates the energetic impact of gluconeogenesis in human metabolism.
Med Hypotheses.
2006; 67(5): 1213–1222. PubMed Abstract
| Publisher Full Text
- 47.
Rydén M, Agustsson T, Laurencikiene J, et al.:
Lipolysis--not inflammation, cell death, or lipogenesis--is involved in adipose tissue loss in cancer cachexia.
Cancer.
2008; 113(7): 1695–1704. PubMed Abstract
| Publisher Full Text
- 48.
Kliewer KL, Ke JY, Tian M, et al.:
Adipose tissue lipolysis and energy metabolism in early cancer cachexia in mice.
Cancer Biol Ther.
2015; 16(6): 886–897. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 49.
Dahlman I, Mejhert N, Linder K, et al.:
Adipose tissue pathways involved in weight loss of cancer cachexia.
Br J Cancer.
2010; 102(10): 1541–1548. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 50.
Tisdale MJ:
Pathogenesis of cancer cachexia.
J Support Oncol.
2003; 1(3): 159–168. PubMed Abstract
- 51.
Das SK, Eder S, Schauer S, et al.:
Adipose triglyceride lipase contributes to cancer-associated cachexia.
Science.
2011; 333(6039): 233–238. PubMed Abstract
| Publisher Full Text
- 52.
Leal Vde O, Mafra D:
Adipokines in obesity.
Clin Chim Acta.
2013; 419: 87–94. PubMed Abstract
| Publisher Full Text
- 53.
Sanders PM, Tisdale MJ:
Effect of zinc-alpha2-glycoprotein (ZAG) on expression of uncoupling proteins in skeletal muscle and adipose tissue.
Cancer Lett.
2004; 212(1): 71–81. PubMed Abstract
| Publisher Full Text
- 54.
Sanchís D, Busquets S, Alvarez B, et al.:
Skeletal muscle UCP2 and UCP3 gene expression in a rat cancer cachexia model.
FEBS Lett.
1998; 436(3): 415–418. PubMed Abstract
| Publisher Full Text
- 55.
Ceperuelo-Mallafre V, Ejarque M, Duran X, et al.:
Zinc-α2-Glycoprotein Modulates AKT-Dependent Insulin Signaling in Human Adipocytes by Activation of the PP2A Phosphatase.
PLoS One.
2015; 10(6): e0129644. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 56.
Purcell SA, Wallengren O, Baracos VE, et al.:
Determinants of change in resting energy expenditure in patients with stage III/IV colorectal cancer.
Clin Nutr.
2020; 39(1): 134–140. PubMed Abstract
| Publisher Full Text
- 57.
Asp ML, Tian M, Wendel AA, et al.:
Evidence for the contribution of insulin resistance to the development of cachexia in tumor-bearing mice.
Int J Cancer.
2010; 126(3): 756–763. PubMed Abstract
| Publisher Full Text
- 58.
Honors MA, Kinzig KP:
The role of insulin resistance in the development of muscle wasting during cancer cachexia.
J Cachexia Sarcopenia Muscle.
2012; 3(1): 5–11. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 59.
Halberg N, Khan T, Trujillo ME, et al.:
Hypoxia-inducible factor 1α induces fibrosis and insulin resistance in white adipose tissue.
Mol Cell Biol.
2009; 29(16): 4467–4483. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 60.
Iiyori N, Alonso LC, Li J, et al.:
Intermittent hypoxia causes insulin resistance in lean mice independent of autonomic activity.
Am J Respir Crit Care Med.
2007; 175(8): 851–857. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 61.
Polotsky VY, Li J, Punjabi NM, et al.:
Intermittent hypoxia increases insulin resistance in genetically obese mice.
J Physiol.
2003; 552(Pt 1): 253–264. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 62.
Camm EJ, Martin-Gronert MS, Wright NL, et al.:
Prenatal hypoxia independent of undernutrition promotes molecular markers of insulin resistance in adult offspring.
FASEB J.
2011; 25(1): 420–427. PubMed Abstract
| Publisher Full Text
- 63.
Cheng N, Cai W, Jiang M, et al.:
Effect of hypoxia on blood glucose, hormones, and insulin receptor functions in newborn calves.
Pediatr Res.
1997; 41(6): 852–856. PubMed Abstract
| Publisher Full Text
- 64.
Warburg O:
On respiratory impairment in cancer cells.
Science.
1956; 124(3215): 269–270. PubMed Abstract
- 65.
Weinhouse S:
Studies on the Fate of Isotopically Labeled Metabolites in the Oxidative Metabolim of Tumors. United States Atomic Energy Commission, Technical Information Service.
Cancer Res.
1951; 11(8): 585–91. PubMed Abstract
- 66.
Solaini G, Sgarbi G, Baracca A:
Oxidative phosphorylation in cancer cells.
Biochim Biophys Acta.
2011; 1807(6): 534–542. PubMed Abstract
| Publisher Full Text
- 67.
Mathupala SP, Ko YH, Pedersen PL:
The pivotal roles of mitochondria in cancer: Warburg and beyond and encouraging prospects for effective therapies.
Biochim Biophys Acta.
2010; 1797(6–7): 1225–1230. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 68.
Smolková K, Plecitá-Hlavatá L, Bellance N, et al.:
Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells.
Int J Biochem Cell Biol.
2011; 43(7): 950–968. PubMed Abstract
| Publisher Full Text
- 69.
Zheng J:
Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review).
Oncol Lett.
2012; 4(6): 1151–1157. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 70.
Lim HY, Ho QS, Low J, et al.:
Respiratory competent mitochondria in human ovarian and peritoneal cancer.
Mitochondrion.
2011; 11(3): 437–443. PubMed Abstract
| Publisher Full Text
- 71.
Scott DA, Richardson AD, Filipp FV, et al.:
Comparative metabolic flux profiling of melanoma cell lines: beyond the warburg effect.
J Biol Chem.
2011; 286(49): 42626–42634. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 72.
Suganuma K, Miwa H, Imai N, et al.:
Energy metabolism of leukemia cells: glycolysis versus oxidative phosphorylation.
Leuk Lymphoma.
2010; 51(11): 2112–2119. PubMed Abstract
| Publisher Full Text
- 73.
Jose C, Bellance N, Rossignol R:
Choosing between glycolysis and oxidative phosphorylation: a tumor's dilemma?
Biochim Biophys Acta.
2011; 1807(6): 552–561. PubMed Abstract
| Publisher Full Text
- 74.
Rodríguez-Enríquez S, Carreño-Fuentes L, Gallardo-Pérez JC, et al.:
Oxidative phosphorylation is impaired by prolonged hypoxia in breast and possibly in cervix carcinoma.
Int J Biochem Cell Biol.
2010; 42(10): 1744–1751. PubMed Abstract
| Publisher Full Text
- 75.
Bellance N, Benard G, Furt F, et al.:
Bioenergetics of lung tumors: alteration of mitochondrial biogenesis and respiratory capacity.
Int J Biochem Cell Biol.
2009; 41(12): 2566–2577. PubMed Abstract
| Publisher Full Text
- 76.
Palmer BF, Clegg DJ:
Ascent to altitude as a weight loss method: the good and bad of hypoxia inducible factor activation.
Obesity (Silver Spring).
2014; 22(2): 311–317. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 77.
Robach P, Cairo G, Gelfi C, et al.:
Strong iron demand during hypoxia-induced erythropoiesis is associated with down-regulation of iron-related proteins and myoglobin in human skeletal muscle.
Blood.
2007; 109(11): 4724–4731. PubMed Abstract
| Publisher Full Text
- 78.
Choi JH, Park MJ, Kim KW, et al.:
Molecular mechanism of hypoxia‐mediated hepatic gluconeogenesis by transcriptional regulation.
FEBS Lett.
2005; 579(13): 2795–2801. PubMed Abstract
| Publisher Full Text
- 79.
Meng F, Zhang W, Wang Y:
RASAL1 inhibits HepG2 cell growth via HIF 2α mediated gluconeogenesis.
Oncol Lett.
2017; 14(6): 7344–7352. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 80.
Suhara T, Hishiki T, Kasahara M, et al.:
Inhibition of the oxygen sensor PHD2 in the liver improves survival in lactic acidosis by activating the Cori cycle.
Proc Natl Acad Sci U S A.
2015; 112(37): 11642–11647. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 81.
Juel C, Halestrap AP:
Lactate transport in skeletal muscle - role and regulation of the monocarboxylate transporter.
J Physiol.
1999; 517(Pt 3): 633–642. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 82.
Carpenter L, Halestrap AP:
The kinetics, substrate and inhibitor specificity of the lactate transporter of Ehrlich-Lettre tumour cells studied with the intracellular pH indicator BCECF.
Biochem J.
1994; 304(Pt 3): 751–760. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 83.
Kaloyianni M, Freedland RA:
Contribution of several amino acids and lactate to gluconeogenesis in hepatocytes isolated from rats fed various diets.
J Nutr.
1990; 120(1): 116–122. PubMed Abstract
| Publisher Full Text
- 84.
Altschuld RA, Kruger FA:
Inhibition of hepatic gluconeogenesis in guinea pig by phenformin.
Ann N Y Acad Sci.
1968; 148(3): 612–622. PubMed Abstract
| Publisher Full Text
- 85.
Hu L, Cui R, Liu H, et al.:
Emodin and rhein decrease levels of hypoxia-inducible factor-1α in human pancreatic cancer cells and attenuate cancer cachexia in athymic mice carrying these cells.
Oncotarget.
2017; 8(50): 88008–88020. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 86.
Dorsey JF, Kao GD:
Aloe(-emodin) for cancer? More than just a comforting salve.
Cancer Biol Ther.
2007; 6(1): 89–90. PubMed Abstract
| Publisher Full Text
- 87.
Srinivas G, Anto RJ, Srinivas P, et al.:
Emodin induces apoptosis of human cervical cancer cells through poly(ADP-ribose) polymerase cleavage and activation of caspase-9.
Eur J Pharmacol.
2003; 473(2-3): 117–125. PubMed Abstract
| Publisher Full Text
- 88.
Shrimali D, Shanmugam MK, Kumar AP, et al.:
Targeted abrogation of diverse signal transduction cascades by emodin for the treatment of inflammatory disorders and cancer.
Cancer Lett.
2013; 341(2): 139–149. PubMed Abstract
| Publisher Full Text
- 89.
Zhang L, Hung MC:
Sensitization of HER-2/neu-overexpressing non-small cell lung cancer cells to chemotherapeutic drugs by tyrosine kinase inhibitor emodin.
Oncogene.
1996; 12(3): 571–576. PubMed Abstract
- 90.
Robinson BH:
Lactic acidemia and mitochondrial disease.
Mol Genet Metab.
2006; 89(1–2): 3–13. PubMed Abstract
| Publisher Full Text
- 91.
Hermansen L, Maehlum S, Pruett EDR, et al.:
Lactate removal at rest and during exercise. In: Metabolic adaptation to prolonged physical exercise. Birkhäuser, Basel. 1975; 101–105. Publisher Full Text
- 92.
van Hall G:
Lactate kinetics in human tissues at rest and during exercise.
Acta Physiol (Oxf).
2010; 199(4): 499–508. PubMed Abstract
| Publisher Full Text
- 93.
Ghezzi P, Dinarello CA, Bianchi M, et al.:
Hypoxia increases production of interleukin-1 and tumor necrosis factor by human mononuclear cells.
Cytokine.
1991; 3(3): 189–194. PubMed Abstract
| Publisher Full Text
- 94.
West MA, Li MH, Seatter SC, et al.:
Pre-exposure to hypoxia or septic stimuli differentially regulates endotoxin release of tumor necrosis factor, interleukin-6, interleukin-1, prostaglandin E2, nitric oxide, and superoxide by macrophages.
J Trauma.
1994; 37(1): 82–9. PubMed Abstract
| Publisher Full Text
- 95.
Albina JE, Henry WL Jr, Mastrofrancesco B, et al.:
Macrophage activation by culture in an anoxic environment.
J Immunol.
1995; 155(9): 4391–4396. PubMed Abstract
- 96.
Metinko AP, Kunkel SL, Standiford TJ, et al.:
Anoxia-hyperoxia induces monocyte-derived interleukin-8.
J Clin Invest.
1992; 90(3): 791–798. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 97.
Scannell G, Waxman K, Kaml GJ, et al.:
Hypoxia induces a human macrophage cell line to release tumor necrosis factor-α and its soluble receptors in vitro.
J Surg Res.
1993; 54(4): 281–285. PubMed Abstract
| Publisher Full Text
- 98.
Koong AC, Chen EY, Giaccia AJ:
Hypoxia causes the activation of nuclear factor κB through the phosphorylation of IκBα on tyrosine residues.
Cancer Res.
1994; 54(6): 1425–1430. PubMed Abstract
- 99.
Taylor CT:
Regulation of intestinal epithelial gene expression in hypoxia.
Kidney Int.
2004; 66(2): 528–531. PubMed Abstract
| Publisher Full Text
- 100.
Taylor CT, Colgan SP:
Therapeutic targets for hypoxia-elicited pathways.
Pharm Res.
1999; 16(10): 1498–1505. PubMed Abstract
| Publisher Full Text
- 101.
Yun JK, McCormick TS, Villabona C, et al.:
Inflammatory mediators are perpetuated in macrophages resistant to apoptosis induced by hypoxia.
Proc Natl Acad Sci U S A.
1997; 94(25): 13903–13908. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 102.
Jeong HJ, Hong SH, Park RK, et al.:
Hypoxia-induced IL-6 production is associated with activation of MAP kinase, HIF-1, and NF-kappaB on HEI-OC1 cells.
Hear Res.
2005; 207(1–2): 59–67. PubMed Abstract
| Publisher Full Text
- 103.
Degrossoli A, Giorgio S:
Functional alterations in macrophages after hypoxia selection.
Exp Biol Med (Maywood).
2007; 232(1): 88–95. PubMed Abstract
- 104.
Rofe AM, Bourgeois CS, Coyle P, et al.:
Altered insulin response to glucose in weight-losing cancer patients.
Anticancer Res.
1994; 14(2B): 647–650. PubMed Abstract
- 105.
Copeland GP, Leinster SJ, Davis JC, et al.:
Insulin resistance in patients with colorectal cancer.
Br J Surg.
1987; 74(11): 1031–1035. PubMed Abstract
| Publisher Full Text
- 106.
Tayek JA:
A review of cancer cachexia and abnormal glucose metabolism in humans with cancer.
J Am Coll Nutr.
1992; 11(4): 445–456. PubMed Abstract
| Publisher Full Text
- 107.
Yoshikawa T, Noguchi Y, Doi C, et al.:
Insulin resistance in patients with cancer: relationships with tumor site, tumor stage, body-weight loss, acute-phase response, and energy expenditure.
Nutrition.
2001; 17(7-8): 590–593. PubMed Abstract
| Publisher Full Text
- 108.
Oltmanns KM, Gehring H, Rudolf S, et al.:
Hypoxia causes glucose intolerance in humans.
Am J Respir Crit Care Med.
2004; 169(11): 1231–1237. PubMed Abstract
| Publisher Full Text
- 109.
Fu C, Jiang L, Zhu F, et al.:
Chronic intermittent hypoxia leads to insulin resistance and impaired glucose tolerance through dysregulation of adipokines in non-obese rats.
Sleep Breath.
2015; 19(4): 1467–1473. PubMed Abstract
| Publisher Full Text
- 110.
Matsumoto S, Yasui H, Mitchell JB, et al.:
Imaging cycling tumor hypoxia.
Cancer Res.
2010; 70(24): 10019–10023. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 111.
Vaupel P:
The role of hypoxia-induced factors in tumor progression.
Oncologist.
2004; 9(suppl_ 5): 10–17. PubMed Abstract
| Publisher Full Text
- 112.
Hockel M, Vaupel P:
Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects.
J Natl Cancer Inst.
2001; 93(4): 266–276. PubMed Abstract
| Publisher Full Text
- 113.
Soeters MR, Soeters PB:
The evolutionary benefit of insulin resistance.
Clin Nutr.
2012; 31(6): 1002–1007. PubMed Abstract
| Publisher Full Text
- 114.
Hatting M, Tavares CD, Sharabi K, et al.:
Insulin regulation of gluconeogenesis.
Ann N Y Acad Sci.
2018; 1411(1): 21. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 115.
Qi C, Pekala PH:
Tumor Necrosis Factor-α–Induced Insulin Resistance in Adipocytes.
Proc Soc Exp Biol Med.
2000; 223(2): 128–135. PubMed Abstract
| Publisher Full Text
- 116.
Hotamisligil GS:
Mechanisms of TNF-α-induced insulin resistance.
Exp Clin Endocrinol Diabetes.
1999; 107(2): 119–125. PubMed Abstract
| Publisher Full Text
- 117.
Hotamisligil GS, Murray DL, Choy LN, et al.:
Tumor necrosis factor alpha inhibits signaling from the insulin receptor.
Proc Natl Acad Sci U S A.
1994; 91(11): 4854–4858. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 118.
Kirwan JP, Hauguel-De Mouzon S, Lepercq J, et al.:
TNF-α is a predictor of insulin resistance in human pregnancy.
Diabetes.
2002; 51(7): 2207–2213. PubMed Abstract
| Publisher Full Text
- 119.
Saghizadeh M, Ong JM, Garvey WT, et al.:
The expression of TNF alpha by human muscle. Relationship to insulin resistance.
J Clin Invest.
1996; 97(4): 1111–1116. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 120.
Noguchi Y, Yoshikawa T, Marat D, et al.:
Insulin resistance in cancer patients is associated with enhanced tumor necrosis factor-α expression in skeletal muscle.
Biochem Biophys Res Commun.
1998; 253(3): 887–892. PubMed Abstract
| Publisher Full Text
- 121.
Rotter V, Nagaev I, Smith U:
Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects.
J Biol Chem.
2003; 278(46): 45777–45784. PubMed Abstract
| Publisher Full Text
- 122.
Briançon‐Marjollet A, Monneret D, Henri M, et al.:
Endothelin regulates intermittent hypoxia‐induced lipolytic remodelling of adipose tissue and phosphorylation of hormone‐sensitive lipase.
J Physiol.
2016; 594(6): 1727–1740. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 123.
Wang GL, Jiang BH, Rue EA, et al.:
Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension.
Proc Natl Acad Sci U S A.
1995; 92(12): 5510–5514. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 124.
Zhong H, De Marzo AM, Laughner E, et al.:
Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases.
Cancer Res.
1999; 59(22): 5830–5835. PubMed Abstract
- 125.
Semenza GL:
Targeting HIF-1 for cancer therapy.
Nat Rev Cancer.
2003; 3(10): 721–732. PubMed Abstract
| Publisher Full Text
- 126.
Iyer, NV, Kotch LE, Agani F, et al.:
Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α.
Genes Dev.
1998; 12(2): 149–162. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 127.
Plotkin BJ, Sigar IM, Swartzendruber JA, et al.:
Differential expression of cytokines and receptor expression during anoxic growth.
BMC Res Notes.
2018; 11(1): 406. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 128.
Gagnon B, Bruera E:
A review of the drug treatment of cachexia associated with cancer.
Drugs.
1998; 55(5): 675–688. PubMed Abstract
| Publisher Full Text
- 129.
Yavuzsen, T, Davis MP, Walsh D, et al.:
Systematic review of the treatment of cancer-associated anorexia and weight loss. In Database of Abstracts of Reviews of Effects (DARE): Quality-assessed Reviews. Centre for Reviews and Dissemination (UK). 2005. Reference Source
- 130.
Gershanovich ML, Danova LA, Ivin BA, et al.:
Results of clinical study of antitumor action of hydrazine sulfate.
Nutr Cancer.
1981; 3(1): 7–12. PubMed Abstract
| Publisher Full Text
- 131.
Integrative PDQ:
Hydrazine Sulfate (PDQ®). In PDQ Cancer Information Summaries. [Internet] National Cancer Institute (US). 2018; Downloaded from Reference Source
- 132.
Simons JP, Aaronson NK, Vansteenkiste JF, et al.:
Effects of Medroxyprogesterone Acetate on Appetite, Weight, and Quality of Life in Advanced-Stage Non-Hormone-Sensitive Cancer: A Placebo-Controlled Multicenter Study.
J Clin Oncol.
1996; 14(4): 1077–1084. PubMed Abstract
| Publisher Full Text
- 133.
Lelli G, Angelelli B, Giambiasi ME:
The anabolic effect of high dose medroxyprogesterone acetate in oncology.
Pharmacol Res Commun.
1983; 15(6): 561–568. PubMed Abstract
| Publisher Full Text
- 134.
Markoe AM:
Role of Medroxyprogesterone in Endocrine-Related Tumors.
Am J Clin Oncol.
1982; 5(4): 457. Publisher Full Text
- 135.
Gebbia V, Testa A, Gebbia N:
Prospective randomised trial of two dose levels of megestrol acetate in the management of anorexia-cachexia syndrome in patients with metastatic cancer.
Br J Cancer.
1996; 73(12): 1576–1580. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 136.
Beller E, Tattersall M, Lumley T:
Improved Quality of Life With Megestrol Acetate in Patients With Endocrine-Insensitive Advanced Cancer: A Randomised Placebo-Controlled Trial. Australasian Megestrol Acetate Cooperative Study Group.
Ann Oncol.
1997; 8(3): 277–283. PubMed Abstract
| Publisher Full Text
- 137.
Feliu J, Gonzalez-Baron M, Berrocal A, et al.:
Usefulness of megestrol acetate in cancer cachexia and anorexia. A placebo-controlled study.
Am J Clin Oncol.
1992; 15(5): 436–440. PubMed Abstract
| Publisher Full Text
- 138.
Davis MP, Feyer P, Ortner P, et al.:
Supportive Oncology E-Book. Elsevier Health Sciences. 2011. Reference Source
- 139.
Whitehouse AS, Smith HJ, Drake JL, et al.:
Mechanism of Attenuation of Skeletal Muscle Protein Catabolism in Cancer Cachexia by Eicosapentaenoic Acid.
Cancer Res.
2001; 61(9): 3604–3609. PubMed Abstract
- 140.
Gorjao R, dos Santos CM, Serdan TD, et al.:
New Insights on the Regulation of Cancer Cachexia by N-3 Polyunsaturated Fatty Acids.
Pharmacol Ther.,
2019; 196: 117–134. PubMed Abstract
| Publisher Full Text
- 141.
Maltoni M, Nanni O, Scarpi E, et al.:
High-dose Progestins for the Treatment of Cancer Anorexia-Cachexia Syndrome: A Systematic Review of Randomised Clinical Trials.
Ann Oncol.
2001; 12(3): 289–300. PubMed Abstract
| Publisher Full Text
- 142.
Argilés JM, López-Soriano FJ, Stemmler B, et al.:
Therapeutic Strategies Against Cancer Cachexia.
Eur J Transl Myol.
2019; 29(1): 7960. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 143.
Ando KTakahashi FMotojima SPossible Role for Tocilizumab, an anti-interleukin-6 Receptor Antibody, in Treating Cancer Cachexia.
J Clin Oncol.
2013; 31(6): e69–e72. PubMed Abstract
| Publisher Full Text
- 144.
Ando K, Takahashi F, Kato M, et al.:
Tocilizumab, a Proposed Therapy for the Cachexia of Interleukin6-expressing Lung Cancer.
PLoS One.
2014; 9(7): e102436. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 145.
Hirata H, Tetsumoto S, Kijima T, et al.:
Favorable Responses to Tocilizumab in Two Patients With Cancer-Related Cachexia.
J Pain Symptom Manage.
2013; 46(2): e9–e13. PubMed Abstract
| Publisher Full Text
- 146.
Lundholm K, Körner U, Gunnebo L, et al.:
Insulin Treatment in Cancer Cachexia: Effects on Survival, Metabolism, and Physical Functioning.
Clin Cancer Res.
2007; 13(9): 2699–2706. PubMed Abstract
| Publisher Full Text
- 147.
Lai V, George J, Richey L, et al.:
Results of a pilot study of the effects of celecoxib on cancer cachexia in patients with cancer of the head, neck, and gastrointestinal tract.
Head Neck.
2008; 30(1): 67–74. PubMed Abstract
| Publisher Full Text
- 148.
Benny Klimek ME, Aydogdu T, Link MJ, et al.:
Acute inhibition of myostatin-family proteins preserves skeletal muscle in mouse models of cancer cachexia.
Biochem Biophys Res Commun.
2010; 391(3): 1548–1554. PubMed Abstract
| Publisher Full Text
- 149.
Langstein HN, Doherty GM, Fraker DL, et al.:
The roles of gamma-interferon and tumor necrosis factor alpha in an experimental rat model of cancer cachexia.
Cancer Res.
1991; 51(9): 2302–2306. PubMed Abstract
- 150.
Tanaka Y, Eda H, Tanaka T, et al.:
Experimental cancer cachexia induced by transplantable colon 26 adenocarcinoma in mice.
Cancer Res.
1990; 50(8): 2290–2295. PubMed Abstract
- 151.
Yan SF, Tritto I, Pinsky D, et al.:
Induction of interleukin 6 (IL-6) by hypoxia in vascular cells. Central role of the binding site for nuclear factor-IL-6.
J Biol Chem.
1995; 270(19): 11463–11471. PubMed Abstract
| Publisher Full Text
- 152.
Hagberg H, Gilland E, Bona E, et al.:
Enhanced expression of interleukin (IL)-1 and IL-6 messenger RNA and bioactive protein after hypoxia-ischemia in neonatal rats.
Pediatr Res.
1996; 40(4): 603–609. PubMed Abstract
| Publisher Full Text
- 153.
Yamauchi-Takihara K, Ihara Y, Ogata A, et al.:
Hypoxic stress induces cardiac myocyte-derived interleukin-6.
Circulation.
1995; 91(5): 1520–1524. PubMed Abstract
| Publisher Full Text
- 154.
Bao B, Ali S, Ahmad A, et al.:
Hypoxia-induced aggressiveness of pancreatic cancer cells is due to increased expression of VEGF, IL-6 and miR-21, which can be attenuated by CDF treatment.
PLoS One.
2012; 7(12): e50165. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 155.
Klausen T, Olsen NV, Poulsen TD, et al.:
Hypoxemia increases serum interleukin-6 in humans.
Eur J Appl Physiol Occup Physiol.
1997; 76(5): 480–482. PubMed Abstract
| Publisher Full Text
- 156.
Bergstrom J:
Mechanisms of uremic suppression of appetite.
J Ren Nutr.
1999; 9(3): 129–132. PubMed Abstract
| Publisher Full Text
- 157.
Harden LM, du Plessis I, Poole S, et al.:
Interleukin (IL)-6 and IL-1beta act synergistically within the brain to induce sickness behavior and fever in rats.
Brain Behav Immun.
2008; 22(6): 838–849. PubMed Abstract
| Publisher Full Text
- 158.
Carrero JJ, Aguilera A, Stenvinkel P, et al.:
Appetite disorders in uremia.
J Ren Nutr.
2008; 18(1): 107–113. PubMed Abstract
| Publisher Full Text
- 159.
Schéle E, Benrick A, Grahnemo L, et al.:
Inter-relation between interleukin (IL)-1, IL-6 and body fat regulating circuits of the hypothalamic arcuate nucleus.
J Neuroendocrinol.
2013; 25(6): 580–589. PubMed Abstract
| Publisher Full Text
- 160.
Hempel SL, Monick MM, Hunninghake GW:
Effect of hypoxia on release of IL-1 and TNF by human alveolar macrophages.
Am J Respir Cell Mol Biol.
1996; 14(2): 170–176. PubMed Abstract
| Publisher Full Text
- 161.
Scannell G:
Leukocyte responses to hypoxic/ischemic conditions.
New horizons (Baltimore, Md.).
1996; 4(2): 179–183. PubMed Abstract
- 162.
Liu F, Liu Y, Lui VC, et al.:
Hypoxia modulates lipopolysaccharide induced TNF-alpha expression in murine macrophages.
Exp Cell Res.
2008; 314(6): 1327–1336. PubMed Abstract
| Publisher Full Text
- 163.
Hung TH, Burton GJ:
Hypoxia and reoxygenation: a possible mechanism for placental oxidative stress in preeclampsia.
Taiwan J Obstet Gynecol.
2006; 45(3): 189–200. PubMed Abstract
| Publisher Full Text
- 164.
Ambler DR, Fletcher NM, Diamond MP, et al.Effects of hypoxia on the expression of inflammatory markers IL-6 and TNF-a in human normal peritoneal and adhesion fibroblasts.
Syst Biol Reprod Med.
2012; 58(6): 324–329. PubMed Abstract
| Publisher Full Text
- 165.
Takabatake N, Nakamura H, Abe S, et al.:
The relationship between chronic hypoxemia and activation of the tumor necrosis factor-alpha system in patients with chronic obstructive pulmonary disease.
Am J Respir Crit Care Med.
2000; 161(4 pt 1): 1179–1184. PubMed Abstract
| Publisher Full Text
- 166.
Kalantar-Zadeh K, Block G, McAllister CJ, et al.:
Appetite and inflammation, nutrition, anemia, and clinical outcome in hemodialysis patients.
Am J Clin Nutr.
2004; 80(2): 299–307. PubMed Abstract
| Publisher Full Text
- 167.
Oner-Iyidogan Y, Gurdol F, Kocak H, et al.:
Appetite-regulating hormones in chronic kidney disease patients.
J Ren Nutr.
2011; 21(4): 316–321. PubMed Abstract
| Publisher Full Text
- 168.
Andréasson A, Arborelius L, Erlanson-Albertsson C, et al.:
A putative role for cytokines in the impaired appetite in depression.
Brain Behav Immun.
2007; 21(2): 147–152. PubMed Abstract
| Publisher Full Text
- 169.
Wilson MM, Philpot C, Morley JE:
Anorexia of aging in long term care: is dronabinol an effective appetite stimulant?--a pilot study.
J Nutr Health Aging.
2007; 11(2): 195–8. PubMed Abstract
- 170.
Zhu L, Wang Q, Zhang L, et al.:
Hypoxia induces PGC-1α expression and mitochondrial biogenesis in the myocardium of TOF patients.
Cell Res.
2010; 20(6): 676–687. PubMed Abstract
| Publisher Full Text
- 171.
Thom R, Rowe GC, Jang C, et al.:
Hypoxic induction of vascular endothelial growth factor (VEGF) and angiogenesis in muscle by truncated peroxisome proliferator-activated receptor γ coactivator (PGC)-1α.
J Biol Chem.
2014; 289(13): 8810–8817. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 172.
Cunningham KF, Beeson GC, Beeson CC, et al.:
Estrogen-Related Receptor α (ERRα) is required for adaptive increases in PGC-1 isoform expression during electrically stimulated contraction of adult cardiomyocytes in sustained hypoxic conditions.
Int J Cardiol.
2015; 187: 393–400. PubMed Abstract
| Publisher Full Text
- 173.
Zu YX, Lu HY, Liu WW, et al.:
Jiang Gui Fang activated interscapular brown adipose tissue and induced epididymal white adipose tissue browning through the PPARγ/SIRT1-PGC1α pathway.
J Ethnopharmacol.
2020; 248: 112271. PubMed Abstract
| Publisher Full Text
- 174.
O'Hagan KA, Cocchiglia S, Zhdanov AV, et al.:
PGC-1alpha is coupled to HIF-1alpha-dependent gene expression by increasing mitochondrial oxygen consumption in skeletal muscle cells.
Proc Natl Acad Sci U S A.
2009; 106(7): 2188–2193. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 175.
Burlington RF, Klain GJ:
Effect of hypoxia on gluconeogenesis in the albino rat and thirteen-lined ground squirrel (Citellus tridecemlineatus).
Comp Biochem Physiol.
1967; 20(1): 275–283. Publisher Full Text
- 176.
Tajima T, Goda N, Fujiki N, et al.:
HIF-1alpha is necessary to support gluconeogenesis during liver regeneration.
Biochem Biophys Res Commun.
2009; 387(4): 789–794. PubMed Abstract
| Publisher Full Text
- 177.
Hardin CD, Roberts TM:
Gluconeogenesis during hypoxia in vascular smooth muscle studied by 13C-NMR.
Physiol Res.
1995; 44(4): 257–260. PubMed Abstract
- 178.
Balsa-Martinez E, Puigserver P:
Cancer Cells Hijack Gluconeogenic Enzymes to Fuel Cell Growth.
Mol Cell.
2015; 60(4): 509–511. PubMed Abstract
| Publisher Full Text
- 179.
Montal ED, Dewi R, Bhalla K, et al.:
PEPCK Coordinates the Regulation of Central Carbon Metabolism to Promote Cancer Cell Growth.
Mol Cell.
2015; 60(4): 571–583. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 180.
Vincent EE, Sergushichev A, Griss T, et al.:
Mitochondrial Phosphoenolpyruvate Carboxykinase Regulates Metabolic Adaptation and Enables Glucose-Independent Tumor Growth.
Mol Cell.
2015; 60(2): 195–207. PubMed Abstract
| Publisher Full Text
- 181.
Azevedo JL Jr, Carey JO, Pories WJ, et al.:
Hypoxia stimulates glucose transport in insulin-resistant human skeletal muscle.
Diabetes.
1995; 44(6): 695–698. PubMed Abstract
| Publisher Full Text
- 182.
Thomas A, Belaidi E, Moulin S, et al.:
Chronic Intermittent Hypoxia Impairs Insulin Sensitivity but Improves Whole-Body Glucose Tolerance by Activating Skeletal Muscle AMPK.
Diabetes.
2017; 66(12): 2942–2951. PubMed Abstract
| Publisher Full Text
- 183.
Gonzalez FJ, Xie C, Jiang C:
The role of hypoxia-inducible factors in metabolic diseases.
Nat Rev Endocrinol.
2019; 15(1): 21–32. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 184.
Priyanka A, Shyni GL, Anupama N, et al.:
Development of insulin resistance through sprouting of inflammatory markers during hypoxia in 3T3-L1 adipocytes and amelioration with curcumin.
Eur J Pharmacol.
2017; 812: 73–81. PubMed Abstract
| Publisher Full Text
- 185.
Bock G, Chittilapilly E, Basu R, et al.:
Contribution of hepatic and extrahepatic insulin resistance to the pathogenesis of impaired fasting glucose: role of increased rates of gluconeogenesis.
Diabetes.
2007; 56(6): 1703–1711. PubMed Abstract
| Publisher Full Text
- 186.
Shulman GI:
Cellular mechanisms of insulin resistance in humans.
Am J Cardiol.
1999; 84(1A): 3J–10J. PubMed Abstract
| Publisher Full Text
- 187.
Zhao W, Li A, Feng X, et al.:
Metformin and resveratrol ameliorate muscle insulin resistance through preventing lipolysis and inflammation in hypoxic adipose tissue.
Cell Signal.
2016; 28(9): 1401–1411. PubMed Abstract
| Publisher Full Text
- 188.
Larsen TS, Myrmel T, Skulberg A, et al.:
Effects of hypoxia on lipolysis in isolated rat myocardial cells.
Mol Cell Biochem.
1989; 88(1–2): 139–144. PubMed Abstract
| Publisher Full Text
- 189.
Drager LF, Jun JC, Polotsky VY:
Metabolic consequences of intermittent hypoxia: relevance to obstructive sleep apnea.
Best Pract Res Clin Endocrinol Metab.
2010; 24(5): 843–851. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 190.
Pasarica M, Rood J, Ravussin E, et al.:
Reduced oxygenation in human obese adipose tissue is associated with impaired insulin suppression of lipolysis.
J Clin Endocrinol Metab.
2010; 95(8): 4052–4055. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 191.
Hashimoto T, Yokokawa T, Endo Y, et al.:
Modest hypoxia significantly reduces triglyceride content and lipid droplet size in 3T3-L1 adipocytes.
Biochem Biophys Res Commun.
2013; 440(1): 43–49. PubMed Abstract
| Publisher Full Text
- 192.
Balaz M, Vician M, Janakova Z, et al.:
Subcutaneous adipose tissue zinc-α2-glycoprotein is associated with adipose tissue and whole-body insulin sensitivity.
Obesity (Silver Spring).
2014; 22(8): 1821–1829. PubMed Abstract
| Publisher Full Text
- 193.
Elattar S, Dimri M, Satyanarayana A:
The tumor secretory factor ZAG promotes white adipose tissue browning and energy wasting.
FASEB J.
2018; 32(9): 4727–4743. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 194.
Elattar S:
The Tumor Secretory Factor ZAG Promotes White Adipose Tissue Browning and Energy Wasting in Cachexia. (Doctoral dissertation, Augusta University). Downoaded from ProQuest Dissertations Publishing, 2018; 10929222.
- 195.
Fan G, Dang X, Li Y, et al.:
Zinc-α2-glycoprotein promotes browning of white adipose tissue in cold-exposed male mice.
Mol Cell Endocrinol.
2020; 501: 110669. PubMed Abstract
| Publisher Full Text
- 196.
Sánchez LM, Chirino AJ, Bjorkman PJ:
Crystal structure of human ZAG, a fat-depleting factor related to MHC molecules.
Science.
1999; 283(5409): 1914–1919. PubMed Abstract
| Publisher Full Text
- 197.
Xiao XH, Qi XY, Wang YD, et al.:
Zinc alpha2 glycoprotein promotes browning in adipocytes.
Biochem Biophys Res Commun.
2018; 496(2): 287–293. PubMed Abstract
| Publisher Full Text
- 198.
Cabassi A, Tedeschi S:
Zinc-α2-glycoprotein as a marker of fat catabolism in humans.
Curr Opin Clin Nutr Metab Care.
2013; 16(3): 267–271. PubMed Abstract
| Publisher Full Text
- 199.
Russell ST, Zimmerman TP, Domin BA, et al.:
Induction of lipolysis in vitro and loss of body fat in vivo by zinc-alpha2-glycoprotein.
Biochim Biophys Acta.
2004; 1636(1): 59–68. PubMed Abstract
| Publisher Full Text
- 200.
Russell ST, Hirai K, Tisdale MJ:
Role of beta3-adrenergic receptors in the action of a tumour lipid mobilizing factor.
Br J Cancer.
2002; 86(3): 424–428. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 201.
Yamashita JI, Ogawa M, Nomura K, et al.:
Interleukin 6 stimulates the production of immunoreactive endothelin 1 in human breast cancer cells.
Cancer Res.
1993; 53(3): 464–467. PubMed Abstract
- 202.
Oikawa T, Kusuhara M, Ishikawa S, et al.:
Production of endothelin-1 and thrombomodulin by human pancreatic cancer cells.
Br J Cancer.
1994; 69(6): 1059–1064. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 203.
Eriksson AKS, Van Harmelen V, Stenson BM, et al.:
Endothelin-1 stimulates human adipocyte lipolysis through the ETA receptor.
Int J Obes (Lond).
2009; 33(1): 67–74. PubMed Abstract
| Publisher Full Text
- 204.
Juan CC, Chang LW, Huang SW, et al.:
Effect of endothelin-1 on lipolysis in rat adipocytes.
Obesity (Silver Spring).
2006; 14(3): 398–404. PubMed Abstract
| Publisher Full Text
- 205.
Davies JC, Bain SC, Kanamarlapudi V:
ADP-ribosylation factor 6 regulates endothelin-1-induced lipolysis in adipocytes.
Biochem Pharmacol.
2014; 90(4): 406–413. PubMed Abstract
| Publisher Full Text
- 206.
Stewart G, Riddick A, Fearon K, et al.:
Proteolysis-inducing factor core peptide is a survival factor in prostate cancer cells subjected to hypoxia and oxidative stress.
2007. Reference Source
- 207.
Wyke SM, Tisdale MJ:
NF-kappaB mediates proteolysis-inducing factor induced protein degradation and expression of the ubiquitin-proteasome system in skeletal muscle.
Br J Cancer.
2005; 92(4): 711–721. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 208.
Todorov PT, Field WN, Tisdale MJ:
Role of a proteolysis-inducing factor (PIF) in cachexia induced by a human melanoma (G361).
Br J Cancer.
1999: 80(11): 1734–1737. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 209.
Lorite MJ, Thompson MG, Drake JL, et al.:
Mechanism of muscle protein degradation induced by a cancer cachectic factor.
Br J Cancer.
1998; 78(7): 850–856. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 210.
Cabal-Manzano R, Bhargava P, Torres-Duarte A, et al.:
Proteolysis-inducing factor is expressed in tumours of patients with gastrointestinal cancers and correlates with weight loss.
Br J Cancer.
2001; 84(12): 1599–1601. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 211.
Watchorn TM, Dowidar N, Dejong CH, et al.:
The cachectic mediator proteolysis inducing factor activates NF-κB and STAT3 in human Kupffer cells and monocytes.
Int J Oncol.
2005; 27(4): 1105–1111. PubMed Abstract
| Publisher Full Text
- 212.
Myrmel T, Forsdahl K, Sager G, et al.:
Regulation of lipolysis in normoxic and hypoxic rat myocytes.
J Mol Cell Cardiol.
1991; 23(2): 207–215. PubMed Abstract
| Publisher Full Text
- 213.
Netzer N, Gatterer H, Faulhaber M, et al.:
Hypoxia, Oxidative Stress and Fat.
Biomolecules.
2015; 5(2): 1143–1150. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 214.
Samuel VT, Shulman GI:
The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux.
J Clin Invest.
2016; 126(1): 12–22. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 215.
Valero T:
Editorial (Thematic Issue: Mitochondrial Biogenesis: Pharmacological Approaches)".
Curr Pharm Des.
2014; 20(35): 5507–9. PubMed Abstract
| Publisher Full Text
- 216.
Sanchis-Gomar F, Garcia-Gimenez JL, Gomez-Cabrera MC, et al.:
Mitochondrial biogenesis in health and disease. Molecular and therapeutic approaches.
Curr Pharm Des.
2014; 20(35): 5619–5633. PubMed Abstract
| Publisher Full Text
- 217.
Summermatter S, Santos G, Pérez-Schindler J, et al.:
Skeletal muscle PGC-1α controls whole-body lactate homeostasis through estrogen-related receptor α-dependent activation of LDH B and repression of LDH A.
Proc Natl Acad Sci U S A.
2013; 110(21): 8738–8743. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 218.
Lehman JJ, Boudina S, Banke NH, et al.:
The transcriptional coactivator PGC-1alpha is essential for maximal and efficient cardiac mitochondrial fatty acid oxidation and lipid homeostasis.
Am J Physiol Heart Circ Physiol.
2008; 295(1): H185–H196. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 219.
Houten SM, Violante S, Ventura FV, et al.:
The biochemistry and physiology of mitochondrial fatty acid β-oxidation and its genetic disorders.
Annu Rev Physiol.
2016; 78: 23–44. PubMed Abstract
| Publisher Full Text
- 220.
Rodgers JT, Lerin C, Haas W, et al.:
Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1.
Nature.
2005; 434(7029): 113–118. PubMed Abstract
| Publisher Full Text
- 221.
Roberts LD, Boström P, O’Sullivan JF, et al.:
β-Aminoisobutyric acid induces browning of white fat and hepatic β-oxidation and is inversely correlated with cardiometabolic risk factors.
Cell Metab.
2014; 19(1): 96–108. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 222.
Ariazi EA, Jordan VC:
Estrogen-related receptors as emerging targets in cancer and metabolic disorders.
Curr Top Med Chem.
2006; 6(3): 203–215. PubMed Abstract
| Publisher Full Text
- 223.
Hentschke M, Susens U, Borgmeyer U:
PGC-1 and PERC, coactivators of the estrogen receptor-related receptor gamma.
Biochem Biophys Res Commun.
2002; 299(5): 872–879. PubMed Abstract
| Publisher Full Text
- 224.
Arany Z, Foo SY, Ma Y, et al.:
HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha.
Nature.
2008; 451(7181): 1008–1012. PubMed Abstract
| Publisher Full Text
- 225.
Soyal SM, Bonova P, Kwik M, et al.:
The Expression of CNS-Specific PPARGC1A Transcripts Is Regulated by Hypoxia and a Variable GT Repeat Polymorphism.
Mol Neurobiol.
2020; 57(2): 752–764. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 226.
Bruns I, Sauer B, Burger MC, et al.:
Disruption of peroxisome proliferator-activated receptor γ coactivator (PGC)-1α reverts key features of the neoplastic phenotype of glioma cells.
J Biol Chem.
2019; 294(9): 3037–3050. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 227.
Rasbach KA, Gupta RK, Ruas JL, et al.:
PGC-1alpha regulates a HIF2alpha-dependent switch in skeletal muscle fiber types.
Proc Natl Acad Sci U S A.
2010; 107(50): 21866–21871. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 228.
Yun CW, Lee JH, Lee SH:
Hypoxia-induced PGC-1α Regulates Mitochondrial Function and Tumorigenesis of Colorectal Cancer Cells.
Anticancer Res.
2019; 39(9): 4865–4876. PubMed Abstract
| Publisher Full Text
- 229.
Vazquez F, Lim JH, Chim H, et al.:
PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress.
Cancer cell.
2013; 23(3): 287–301. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 230.
Haq R, Shoag J, Andreu-Perez P, et al.:
Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF.
Cancer cell.
2013; 23(3): 302–315. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 231.
Lei Q, Qiang F, Chao D, et al.:
Amelioration of hypoxia and LPS-induced intestinal epithelial barrier dysfunction by emodin through the suppression of the NF-κB and HIF-1α signaling pathways.
Int J Mol Med.
2014; 34(6): 1629–1639. PubMed Abstract
| Publisher Full Text
- 232.
Wu J, Ke X, Wang W, et al.:
Aloe-emodin suppresses hypoxia-induced retinal angiogenesis via inhibition of HIF-1α/VEGF pathway.
Int J Biol Sci.
2016; 12(11): 1363–1371. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 233.
Hsu SC, Chung JG:
Anticancer potential of emodin.
BioMedicine.
2012; 2(3): 108–116. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 234.
Hwang SY, Heo K, Kim JS, et al.:
Emodin attenuates radioresistance induced by hypoxia in HepG2 cells via the enhancement of PARP1 cleavage and inhibition of JMJD2B.
Oncol Rep.
2015; 33(4): 1691–1698. PubMed Abstract
| Publisher Full Text
- 235.
Liu A, Chen H, Tong H, et al.:
Emodin potentiates the antitumor effects of gemcitabine in pancreatic cancer cells via inhibition of nuclear factor-κB.
Mol Med Rep.
2011; 4(2): 221–227. PubMed Abstract
| Publisher Full Text
- 236.
Agbenyega T, Planche T, Bedu-Addo G, et al.:
Population kinetics, efficacy, and safety of dichloroacetate for lactic acidosis due to severe malaria in children.
J Clin Pharmacol.
2003; 43(4): 386–396. PubMed Abstract
| Publisher Full Text
- 237.
Chu PI:
Pharmacokinetics of Sodium Dichloroacetate. PhD dissertation. Gainesville, FL:University of Florida. 1987. Reference Source
- 238.
Stacpoole PW:
The pharmacology of dichloroacetate.
Metabolism.
1989; 38(11): 1124–1144. PubMed Abstract
| Publisher Full Text
- 239.
Shroads AL, Guo X, Dixit V, et al.:
Age-dependent kinetics and metabolism of dichloroacetate: possible relevance to toxicity.
J Pharmacol Exp Ther.
2008; 324(3): 1163–1171. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 240.
Wells PG, Moore GW, Rabin D, et al.:
Metabolic effects and pharmacokinetics of intravenously administered dichloroacetate in humans.
Diabetologia.
1980; 19(2): 109–113. PubMed Abstract
| Publisher Full Text
- 241.
Stacpoole PW, Moore GW, Kornhauser DM:
Metabolic effects of dichloroacetate in patients with diabetes mellitus and hyperlipoproteinemia.
N Engl J Med.
1978; 298(10): 526–530. PubMed Abstract
| Publisher Full Text
- 242.
Gonzalez-Leon A, Schultz IR, Xu G, et al.:
Pharmacokinetics and metabolism of dichloroacetate in the F344 rat after prior administration in drinking water.
Toxicol Appl Pharmacol.
1997; 146(2): 189–195. PubMed Abstract
| Publisher Full Text
- 243.
Lukas G, Vyas KH, Brindle SD, et al.:
Biological disposition of sodium dichloroacetate in animals and humans after intravenous administration.
J Pharm Sci.
1980; 69(4): 419–21. PubMed Abstract
| Publisher Full Text
- 244.
Maisenbacher HW 3rd, Shroads AL 3rd, Zhong G, et al.:
Pharmacokinetics of oral dichloroacetate in dogs.
J Biochem Mol Toxicol.
2013; 27(12): 522–525. PubMed Abstract
| Publisher Full Text
- 245.
Stacpoole PW, Henderson GN, Yan Z, et al.:
Clinical pharmacology and toxicology of dichloroacetate.
Environ Health Perspect.
1998; 106 Suppl 4: 989–94. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 246.
Williams PJ, Lane JR, Turkel CC, et al.:
Dichloroacetate: population pharmacokinetics with a pharmacodynamic sequential link model.
J Clin Pharmacol.
2001; 41(3): 259–267. PubMed Abstract
| Publisher Full Text
- 247.
Michelakis ED, Webster L, Mackey JR:
Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer.
Br J Cancer.
2008; 99(7): 989–94. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 248.
Stockwin LH, Yu SX, Borgel S, et al.:
Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC.
Int J Cancer.
2010; 127(11): 2510–2519. PubMed Abstract
| Publisher Full Text
- 249.
Zell R, Geck P, Werdan K, et al.:
TNF-alpha and IL-1 alpha inhibit both pyruvate dehydrogenase activity and mitochondrial function in cardiomyocytes: evidence for primary impairment of mitochondrial function.
Mol Cell Biochem.
1997; 177(1–2): 61–67. PubMed Abstract
| Publisher Full Text
- 250.
Sutendra G, Dromparis P, Bonnet S, et al.:
Pyruvate dehydrogenase inhibition by the inflammatory cytokine TNFα contributes to the pathogenesis of pulmonary arterial hypertension.
J Mol Med (Berl).
2011; 89(8): 771–83. PubMed Abstract
| Publisher Full Text
- 251.
Wang P, Chen M, Yang Z, et al.:
Activation of Pyruvate Dehydrogenase Activity by Dichloroacetate Improves Survival and Neurologic Outcomes After Cardiac Arrest in Rats.
Shock.
2018; 49(6): 704–711. PubMed Abstract
| Publisher Full Text
- 252.
Kumar A, Kant S, Singh SM:
Novel molecular mechanisms of antitumor action of dichloroacetate against T cell lymphoma: Implication of altered glucose metabolism, pH homeostasis and cell survival regulation.
Chem Biol Interact.
2012; 199(1): 29–37. PubMed Abstract
| Publisher Full Text
- 253.
Zentella A, Manogue K, Cerami A:
Cachectin/TNF-mediated lactate production in cultured myocytes is linked to activation of a futile substrate cycle.
Cytokine.
1993; 5(5): 436–447. PubMed Abstract
| Publisher Full Text
- 254.
Diamond MP, Suhrer JH Jr, Williams PE, et al.:
Dichloroacetate: direct inhibitor of lipolysis in the conscious dog.
Horm Metab Res.
1982; 14(2): 102–103. PubMed Abstract
| Publisher Full Text
- 255.
Blackshear PJ, Holloway PA, Albert KG:
The metabolic effects of sodium dichloroacetate in the starved rat.
Biochem J.
1974; 142(2): 279–286. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 256.
Constantin-Teodosiu D:
Regulation of muscle pyruvate dehydrogenase complex in insulin resistance: effects of exercise and dichloroacetate.
Diabetes Metab J.
2013; 37(5): 301–314. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 257.
Whitehouse S, Cooper RH, Randle PJ:
Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids.
Biochem J.
1974; 141(3): 761–774. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 258.
Harris RA, Crabb DW:
Inhibition of hepatic gluconeogenesis by dichloroacetate.
Arch Biochem Biophys.
1978; 189(2): 364–371. PubMed Abstract
| Publisher Full Text
- 259.
Michelakis ED, Sutendra G, Dromparis P, et al.:
Metabolic modulation of glioblastoma with dichloroacetate.
Sci Transl Med.
2010; 2(31): 31ra34. PubMed Abstract
| Publisher Full Text
- 260.
Kinnaird A, Dromparis P, Saleme B, et al.:
Metabolic Modulation of Clear-cell Renal Cell Carcinoma with Dichloroacetate, an Inhibitor of Pyruvate Dehydrogenase Kinase.
Eur Urol.
2016; 69(4): 734–744. PubMed Abstract
| Publisher Full Text
- 261.
Dhar S, Lippard SJ:
Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate.
Proc Natl Acad Sci U S A.
2009; 106(52): 22199–22204. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 262.
Xie J, Wang BS, Yu DH, et al.:
Dichloroacetate shifts the metabolism from glycolysis to glucose oxidation and exhibits synergistic growth inhibition with cisplatin in HeLa cells.
Int J Oncol.
2011; 38(2): 409–417. PubMed Abstract
| Publisher Full Text
- 263.
Zeng S, Liang H, Guan G:
Dichloroacetate enhances the cytotoxic effect of Cisplatin via decreasing the level of FOXM1 in prostate cancer.
Int J Clin Med.
2016; 9: 11044–11050. Reference Source
- 264.
Wang M, Liao C, Hu Y, et al.:
Sensitization of breast cancer cells to paclitaxel by dichloroacetate through inhibiting autophagy.
Biochem Biophys Res Commun.
2017; 489(2): 103–108. PubMed Abstract
| Publisher Full Text
- 265.
Lu X, Zhou D, Hou B, et al.:
Dichloroacetate enhances the antitumor efficacy of chemotherapeutic agents via inhibiting autophagy in non-small-cell lung cancer.
Cancer Manag Res.
2018; 10: 1231–1241. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 266.
Sun H, Zhu A, Zhou X, et al.:
Suppression of pyruvate dehydrogenase kinase-2 re-sensitizes paclitaxel-resistant human lung cancer cells to paclitaxel.
Oncotarget.
2017; 8(32): 52642–52650. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 267.
Kolesnik DL, Pyaskovskaya ON, Yakshibaeva YR, et al.:
Time-dependent cytotoxicity of dichloroacetate and metformin against Lewis lung carcinoma.
Exp Oncol.
2019; 41(1): 14–19. PubMed Abstract
| Publisher Full Text
- 268.
Inanc S, Keles D, Eskiizmir G, et al.:
Metformin And Dichloroacetate Combination Exert A Synergistic Effect On Cell Viability Of Oral Squamous Cell Carcinoma.
ENT Updates.
2019; 9(2): 68–73. Publisher Full Text
- 269.
Kolesnik DL, Pyaskovskaya ON, Yurchenko OV, et al.:
Metformin enhances antitumor action of sodium dichloroacetate against glioma C6.
Exp Oncol.
2019; 41(2): 123–129. PubMed Abstract
| Publisher Full Text
- 270.
Jianping D, Lan QU, Wang FE, et al.:
Metformin and Dichloroacetate Cocrystals Suppress the Growth of Triple-Negative Breast Cancer.
Lat Am J Pharm.
2019; 38(9): 1754–1762. Reference Source
- 271.
Prokhorova IV, Pyaskovskaya ON, Kolesnik DL, et al.:
Influence of metformin, sodium dichloroacetate and their combination on the hematological and biochemical blood parameters of rats with gliomas C6.
Exp Oncol.
2018; 40(3): 205–210. PubMed Abstract
| Publisher Full Text
- 272.
Voltan R, Rimondi E, Melloni E, et al.:
Metformin combined with sodium dichloroacetate promotes B leukemic cell death by suppressing anti-apoptotic protein Mcl-1.
Oncotarget.
2016; 7(14): 18965–77. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 273.
Haugrud AB, Zhuang Y, Coppock JD, et al.:
Dichloroacetate enhances apoptotic cell death via oxidative damage and attenuates lactate production in metformin-treated breast cancer cells.
Breast Cancer Res Treat.
2014; 147(3): 539–550. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 274.
Kiliccioglu I, Konac E, Albayrak G, et al.:
Combination of Metformin and Dichloroacetate Inhibits Proliferation and Induce Intrinsic Pathway of Apoptosis in PC-3 Human Prostate Cancer Cells.
Gazi Medical Journal.
2015; 26(4A). Publisher Full Text
- 275.
Dezube BJ, Sherman ML, Fridovich-Keil JL, et al.:
Down-regulation of tumor necrosis factor expression by pentoxifylline in cancer patients: a pilot study.
Cancer Immunol Immunother.
1993; 36(1): 57–60. PubMed Abstract
| Publisher Full Text
- 276.
Dezube B, Fridovich-Keil J, Bouvard I, et al.:
Pentoxifylline and wellbeing in patients with cancer.
Lancet.
1990; 335(8690): 662. PubMed Abstract
| Publisher Full Text
- 277.
Lilly CM, Sandhu JS, Ishizaka A, et al.:
Pentoxifylline prevents tumor necrosis factor-induced lung injury.
Am Rev Respir Dis.
1989; 139(6): 1361–1368. PubMed Abstract
| Publisher Full Text
- 278.
Noel P, Nelson S, Bokulic R, et al.:
Pentoxifylline inhibits lipopolysaccharide-induced serum tumor necrosis factor and mortality.
Life Sci.
1990; 47(12): 1023–1029. PubMed Abstract
| Publisher Full Text
- 279.
Strieter RM, Remick DG, Ward PA, et al.:
Cellular and molecular regulation of tumor necrosis factor-alpha production by pentoxifylline.
Biochem Biophys Res Commun.
1988; 155(3): 1230–1236. PubMed Abstract
| Publisher Full Text
- 280.
Sampaio EP, Moraes MO, Nery JA, et al.:
Pentoxifylline decreases in vivo and in vitro tumour necrosis factor-alpha (TNF-alpha) production in lepromatous leprosy patients with erythema nodosum leprosum (ENL).
Clin Exp Immunol.
1998; 111(2): 300–8. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 281.
Satapathy SK, Garg S, Chauhan R, et al.:
Beneficial effects of tumor necrosis factor-alpha inhibition by pentoxifylline on clinical, biochemical, and metabolic parameters of patients with nonalcoholic steatohepatitis.
Am J Gastroenterol.
2004; 99(10): 1946–1952. PubMed Abstract
- 282.
Ward A, Clissold SP:
Pentoxifylline. A review of its pharmacodynamic and pharmacokinetic properties, and its therapeutic efficacy.
Drugs.
1987; 34(1): 50–97. PubMed Abstract
| Publisher Full Text
- 283.
Goldberg RM, Loprinzi CL, Mailliard JA, et al.:
Pentoxifylline for treatment of cancer anorexia and cachexia? A randomized, double-blind, placebo-controlled trial.
J Clin Oncol.
1995; 13(11): 2856–2859. PubMed Abstract
| Publisher Full Text
- 284.
Zywietz F, Böhm L, Sagowski C, et al.:
Pentoxifylline enhances tumor oxygenation and radiosensitivity in rat rhabdomyosarcomas during continuous hyperfractionated irradiation.
Strahlenther Onkol.
2004; 180(5): 306–314. PubMed Abstract
| Publisher Full Text
- 285.
Lee I, Boucher Y, Demhartner TJ, et al.:
Changes in tumour blood flow, oxygenation and interstitial fluid pressure induced by pentoxifylline.
Br J Cancer.
1994; 69(3): 492–496. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 286.
Mateo J, García-Lecea M, Cadenas S, et al.:
Regulation of hypoxia-inducible factor-1alpha by nitric oxide through mitochondria-dependent and -independent pathways.
Biochem J.
2003; 376(Pt 2): 537–544. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 287.
Sogawa K, Numayama-Tsuruta K, Ema M, et al.:
Inhibition of hypoxia-inducible factor 1 activity by nitric oxide donors in hypoxia.
Proc Natl Acad Sci U S A.
1998; 95(13): 7368–7373. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 288.
Burke AJ, Sullivan FJ, Giles FJ, et al.:
The yin and yang of nitric oxide in cancer progression.
Carcinogenesis.
2013; 34(3): 503–512. PubMed Abstract
| Publisher Full Text
- 289.
Sukhatme V, Bouche G, Meheus L, et al.:
Repurposing Drugs in Oncology (ReDO)-nitroglycerin as an anti-cancer agent.
Ecancermedicalscience.
2015; 9: 568. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 290.
Tavares JL, Wangoo A, Dilworth P, et al.:
Thalidomide reduces tumor necrosis factor-alpha production by human alveolar macrophages.
Respir Med.
1997; 91(1): 31–9. PubMed Abstract
| Publisher Full Text
- 291.
Bruera E, Neumann CM, Pituskin E, et al.:
Thalidomide in patients with cachexia due to terminal cancer: preliminary report.
Ann Oncol.
1999; 10(7): 857–859. PubMed Abstract
| Publisher Full Text
- 292.
Tramontana JM, Utaipat U, Molloy A, et al.:
Thalidomide treatment reduces tumor necrosis factor alpha production and enhances weight gain in patients with pulmonary tuberculosis.
Mol Med.
1995; 1(4): 384–397. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 293.
Reyes-Terán G, Sierra-Madero JG, Martínez del Cerro V, et al.:
Effects of thalidomide on HIV-associated wasting syndrome: a randomized, double-blind, placebo-controlled clinical trial.
AIDS.
1996; 10(13): 1501–1507. PubMed Abstract
| Publisher Full Text
- 294.
Kumar S, Witzig TE, Rajkumar SV:
Thalidomide as an anti-cancer agent.
J Cell Mol Med.
2002; 6(2): 160–174. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 295.
Vergara TRC, Samer S, Santos-Oliveira JR, et al.:
Thalidomide is Associated With Increased T Cell Activation and Inflammation in Antiretroviral-naive HIV-infected Individuals in a Randomised Clinical Trial of Efficacy and Safety.
EBioMedicine.
2017; 23: 59–67. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 296.
Luzzio FA:
Thalidomide and Analogues. In
Imides.
Elsevier. 2019; 367–429. Publisher Full Text
- 297.
Ma F, Hu L, Yu M, et al.:
Emodin Decreases Hepatic Hypoxia-Inducible Factor-1 α by Inhibiting its Biosynthesis.
Am J Chin Med.
2016; 44(5): 997–1008. PubMed Abstract
| Publisher Full Text
- 298.
Ha MK, Song YH, Jeong SJ, et al.:
Emodin inhibits proinflammatory responses and inactivates histone deacetylase 1 in hypoxic rheumatoid synoviocytes.
Biol Pharm Bull.
2011; 34(9): 1432–1437. PubMed Abstract
| Publisher Full Text
- 299.
Xing YX, Li MH, Tao L, et al.:
Anti-Cancer Effects of Emodin on HepG2 Cells as Revealed by 1H NMR Based Metabolic Profiling.
J Proteome Res.
2018; 17(5): 1943–1952. PubMed Abstract
| Publisher Full Text
- 300.
Lu Y, Jeong YT, Li X, et al.:
Emodin Isolated from Polygoni cuspidati Radix Inhibits TNF-α and IL-6 Release by Blockading NF-κB and MAP Kinase Pathways in Mast Cells Stimulated with PMA Plus A23187.
Biomol Ther (Seoul).
2013; 21(6): 435–41. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 301.
Gupta SC, Rai V:
Role of Emodin in Chemosensitization of Cancer. In
Role of Nutraceuticals in Cancer Chemosensitization.
Academic Press. 2018; 241–257. Publisher Full Text
- 302.
Chen S, Zhang Z, Zhang J:
Emodin enhances antitumor effect of paclitaxel on human non-small-cell lung cancer cells in vitro and in vivo.
Drug Des Devel Ther.
2019; 13: 1145–1153. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 303.
Ko JC, Su YJ, Lin ST, et al.:
Emodin enhances cisplatin-induced cytotoxicity via down-regulation of ERCC1 and inactivation of ERK1/2.
Lung Cancer.
2010; 69(2): 155–164. PubMed Abstract
| Publisher Full Text
- 304.
Wang J, Yi J:
Cancer cell killing via ROS: to increase or decrease, that is the question.
Cancer Biol Ther.
2008; 7(12): 1875–1884. PubMed Abstract
| Publisher Full Text
- 305.
Lin SZ, Wei WT, Chen H, et al.:
Antitumor activity of emodin against pancreatic cancer depends on its dual role: promotion of apoptosis and suppression of angiogenesis.
PLoS One.
2012; 7(8): e42146. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 306.
Kaneshiro T, Morioka T, Inamine M, et al.:
Anthraquinone derivative emodin inhibits tumor-associated angiogenesis through inhibition of extracellular signal-regulated kinase 1/2 phosphorylation.
Eur J Pharmacol.
2006; 553(1–3): 46–53. PubMed Abstract
| Publisher Full Text
- 307.
Ma J, Lu H, Wang S, et al.:
The anthraquinone derivative Emodin inhibits angiogenesis and metastasis through downregulating Runx2 activity in breast cancer.
Int J Oncol.
2015; 46(4): 1619–1628. PubMed Abstract
| Publisher Full Text
- 308.
Zheng Q, Xu Y, Lu J, et al.:
Emodin inhibits migration and invasion of human endometrial stromal cells by facilitating the mesenchymal–epithelial transition through targeting ILK.
Reprod Sci.
2016; 23(11): 1526–1535. PubMed Abstract
| Publisher Full Text
- 309.
Yan YY, Zheng LS, Zhang X, et al.:
Blockade of Her2/neu Binding to Hsp90 by Emodin Azide Methyl Anthraquinone Derivative Induces Proteasomal Degradation of Her2/neu.
Mol Pharm.
2011; 8(5): 1687–1697. PubMed Abstract
| Publisher Full Text
- 310.
Koerner SK, Hanai JI, Bai S, et al.:
Design and synthesis of emodin derivatives as novel inhibitors of ATP-citrate lyase.
Eur J Med Chem.
2017; 126: 920–928. PubMed Abstract
| Publisher Full Text
- 311.
Tang Q, Wu J, Zheng F, et al.:
Emodin increases expression of insulin-like growth factor binding protein 1 through activation of MEK/ERK/AMPKα and interaction of PPARγ and Sp1 in lung cancer.
Cell Physiol Biochem.
2017; 41(1): 339–357. PubMed Abstract
| Publisher Full Text
- 312.
Chun-ming QI, Hua-xin HO, Dao-hai CH, et al.:
The Reversal Effect of Emodin on Cisplatin Resistance in Ovarian Cancer Cells and the Expression of Resistance-associated Genes.
Natural Product Research & Development.
2011; 23(4).
- 313.
Subramaniam A, Shanmugam MK, Ong TH, et al.:
Emodin inhibits growth and induces apoptosis in an orthotopic hepatocellular carcinoma model by blocking activation of STAT3.
Br J Pharmacol.
2013; 170(4): 807–821. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 314.
Dong X, Zeng Y, Liu Y, et al.:
Aloe-emodin: A review of its pharmacology, toxicity, and pharmacokinetics.
Phytother Res.
2020; 34(2): 270–281. PubMed Abstract
| Publisher Full Text
- 315.
Yuan L, Ziegler R, Hamann A:
Inhibition of phosphoenolpyruvate carboxykinase gene expression by metformin in cultured hepatocytes.
Chin Med J (Engl).
2002; 115(12): 1843–1848. PubMed Abstract
- 316.
Davies GF, Khandelwal RL, Wu L, et al.:
Inhibition of phosphoenolpyruvate carboxykinase (PEPCK) gene expression by troglitazone: a peroxisome proliferator-activated receptor-gamma (PPARgamma)-independent, antioxidant-related mechanism.
Biochem Pharmacol.
2001; 62(8): 1071–1079. PubMed Abstract
| Publisher Full Text
- 317.
Xia X, Yan J, Shen Y, et al.:
Berberine improves glucose metabolism in diabetic rats by inhibition of hepatic gluconeogenesis.
PLoS One.
2011; 6(2): e16556. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 318.
Yang X, Yang B, Cai J, et al.:
Berberine enhances radiosensitivity of esophageal squamous cancer by targeting HIF-1α in vitro and in vivo.
Cancer Biol Ther.
2013; 14(11): 1068–1073. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 319.
Guamán Ortiz LM, Lombardi P, Tillhon M, et al.:
Berberine, an Epiphany Against Cancer.
Molecules.
2014; 19(8): 12349–12367. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 320.
Fukuda K, Hibiya Y, Mutoh M, et al.:
Inhibition by berberine of cyclooxygenase-2 transcriptional activity in human colon cancer cells.
J Ethnopharmacol.
1999; 66(2): 227–233. PubMed Abstract
| Publisher Full Text
- 321.
Meeran SM, Katiyar S, Katiyar SK:
Berberine-induced apoptosis in human prostate cancer cells is initiated by reactive oxygen species generation.
Toxicol Appl Pharmacol.
2008; 229(1): 33–43. PubMed Abstract
| Publisher Full Text
- 322.
Patil JB, Kim J, Jayaprakasha GK:
Berberine Induces Apoptosis in Breast Cancer Cells (MCF-7) Through Mitochondrial-Dependent Pathway.
Eur J Pharmacol.
2010; 645(1–3): 70–78. PubMed Abstract
| Publisher Full Text
- 323.
Kaboli PJ, Rahmat A, Ismail P, et al.:
Targets and Mechanisms of Berberine, a Natural Drug With Potential to Treat Cancer With Special Focus on Breast Cancer.
Eur J Pharmacol.
2014; 740: 584–595. PubMed Abstract
| Publisher Full Text
- 324.
Fan T, Sun G, Sun X, et al.:
Tumor Energy Metabolism and Potential of 3-Bromopyruvate as an Inhibitor of Aerobic Glycolysis: Implications in Tumor Treatment.
Cancers (Basel).
2019; 11(3): pii: E317. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 325.
Darabedian N, Chen TC, Molina H, et al.:
Bioorthogonal Profiling of a Cancer Cell Proteome Identifies a Large Set of 3-Bromopyruvate Targets Beyond Glycolysis.
ACS Chem Biol.
2018; 13(11): 3054–3058. PubMed Abstract
| Publisher Full Text
- 326.
Azevedo-Silva, J, Queirós, O, Baltazar, F, et al.:
The anticancer agent 3-bromopyruvate: a simple but powerful molecule taken from the lab to the bedside.
J Bioenerg Biomembr.
2016; 48(4): 349–362. PubMed Abstract
| Publisher Full Text
- 327.
Oldfield V, Dhillon S, Plosker GL, et al.:
Tocilizumab.
Drugs.
2009; 69(5): 609–632. PubMed Abstract
| Publisher Full Text
- 328.
Narsale AA, Carson JA:
Role of IL-6 in cachexia–therapeutic implications.
Curr Opin Support Palliat Care.
2014; 8(4): 321–7. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 329.
Berti A, Boccalatte F, Sabbadini MG, et al.:
Assessment of tocilizumab in the treatment of cancer cachexia.
J Clin Oncol.
2013; 31(23): 2970. PubMed Abstract
| Publisher Full Text
- 330.
Mitsunaga S, Okusaka T, Ikeda M, et al.:
Multicenter, open-Label, phase I/II study of tocilizumab, an anti-Interleukin-6 receptor monoclonal antibody, combined with gemcitabine in patients with advanced pancreatic cancer.
J Med Diagn Methods.
2017; 6: 1–6. Publisher Full Text
- 331.
Nishino N, Tamori Y, Kasuga M:
Insulin efficiently stores triglycerides in adipocytes by inhibiting lipolysis and repressing PGC-1alpha induction.
Kobe J Med Sci.
2007; 53(3): 99–106. PubMed Abstract
- 332.
Hamauchi S, Furuse J, Takano T, et al.:
A multicenter, open-label, single-arm study of anamorelin (ONO-7643) in advanced gastrointestinal cancer patients with cancer cachexia.
Cancer.
2019; 125(23): 4294–4302. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 333.
William MA, Friend J, Polvino W, et al.: U.S. Patent No. 10,278,964. Washington, DC U.S. Patent and Trademark Office. 2019. Reference Source
- 334.
Naito T:
Emerging Treatment Options For Cancer-Associated Cachexia: A Literature Review.
Ther Clin Risk Manag.
2019; 15: 1253–1266. PubMed Abstract
| Publisher Full Text
| Free Full Text
- 335.
https://www.ema.europa.eu/en/documents/smop-initial/questions-answers-refusal-marketing-authorisation-adlumiz-anamorelin-hydrochloride_en.pdf
- 336.
Petruzzelli M, Schweiger M, Schreiber R, et al.:
A switch from white to brown fat increases energy expenditure in cancer-associated cachexia.
Cell Metab.
2014; 20(3): 433–447. PubMed Abstract
| Publisher Full Text
- 337.
Blum D, Omlin A, Baracos VE, et al.:
Cancer cachexia: a systematic literature review of items and domains associated with involuntary weight loss in cancer.
Crit Rev Oncol Hematol.
2011; 80(1): 114–144. PubMed Abstract
| Publisher Full Text
Comments on this article Comments (0)