Keywords
RNA Editing Sites, Sequence Based Features, Feature Selection, Incremental Decision Tree
RNA Editing Sites, Sequence Based Features, Feature Selection, Incremental Decision Tree
RNA editing is a process by which insertion, deletion or substitution of base nucleotides happen within the RNA. Such editing changes occur within the cell meaning nucleotide sequences are altered. This may cause alteration in the peptide sequence, which could be entirely different from that encoded in the DNA. Adenosine to Inosine (A-to-I) is the most common of all RNA editing events. Apart from making genetic alterations and resulting in codon changes A-to-I is often responsible for several intracellular operations like alternative splicing1. A-to-I is found to have its effect on RNA secondary structural changes2. Moreover, it is found to be in correlation with cancer formation3. Thus, research and identification of RNA editing is very important.
Experimental methods like RNA-Seq4 are effective means for identification of A-to-I sites. However, they are time consuming. Moreover, as known RNA-editing sites are available, computational methods for prediction of RNA editing sites are becoming more relevant in this era of knowledge and data discovery. Various computational methods are proposed in the literature for prediction of RNA editing sites, including Genetic Algorithms (GA)5, Support Vector Machines (SVM)6, Logistic Regression (LR)7 and Deep Learning8. PREPACT was proposed as a computational tool for C-to-U and U-to-C RNA editing site prediction for plant species9. Another method was presented by 10 for prediction of C-to-U editing sites using biochemical and evolutionary information.
Several machine learning based prediction methods are proposed in the literature of A-to-I RNA editing site identification in recent years. PAI was proposed by 11 that used pseudo nucleotide compositions as features with a SVM classification algorithm. They proposed and built two benchmark datasets derived from the Drosophila genome based on the work by 12. In a subsequent work, they proposed iRNA-AI6 using general form of pseudo nucleotide composition (PseKNC) with SVM and further improved the previous results on A-to-I datasets. A prediction method iRNA-3typeA13 was proposed to detect and identify three types of RNA-modifications at Adenosine sites. In a recent work, auto-encoders were used to develop PAI-SAE8 for prediction of A-to-I sites.
In this paper, we propose and present PRESa2i, a novel prediction method for A-to-I RNA Editing sites. In our proposed method, we have used the Hoeffding tree, an incremental decision tree for classification of samples. We have used sequence based features extracted from RNA samples collected as positive and negative instances in the dataset. We have used a novel feature selection method to select only the top 179 features. On a standard benchmark dataset PRESa2i has achieved a significant accuracy of 86.48% and on the independent test set it has a significant sensitivity of 90.67%. We have also made our method freely available for use by researchers at: http://brl.uiu.ac.bd/presa2i/index.php.
PRESa2i is trained with a dataset that has sequence based features. A novel feature selection algorithm is applied to select the top features and incremental decision tree algorithms are learned. The model is built on a standard benchmark training set and the performance of the model is tested using an independent set. As suggested by 14 and followed by many researchers15–18, we follow five steps for establishing any good tool for attribute prediction of biological entities: i) selection of benchmark datasets, ii) feature generation, iii) selection of appropriate algorithm, iv) evaluation methods, and v) establishment of a web server.
The first step in any machine learning based supervised prediction task is to select a standard set of data. The RNA A-to-I editing site prediction problem is formulated as a binary classification problem, where Adenine in a RNA sequence or subsequence is labeled with two classes: A-to-I or positive samples and non A-to-I or negative samples. A dataset can be formally defined as the following:
Here, denotes the dataset and positive and negative datasets are denoted by and respectively. The datasets that we have used is first used in 11 based on the work in 12. The original dataset constructed in 12 contained 127 A-to-I editing sites with sequences and 127 non A-to-I editing sites with sequences. These sequences were obtained by sequencing wild-type and ADAR-deficient D. melanogaster DNA and RNA. After removing the redundant sequences, a benchmark dataset was constructed that contained 125 positive or A-to-I sites with sequences and 119 negative or non A-to-I sequence sites. Each of these RNA sequences in the dataset are 51 nucleotides long. From herein, we refer to this dataset as benchmark dataset.
An independent test set was constructed by further analyzing the sequences of D. melanogaster by 19. This set contained 51 nucleotide long 300 positive A-to-I sites with sequence. A summary of the datasets are presented in Table 1. Note that the benchmark dataset is balanced, while the independent dataset contains only positive sequences. Both datasets used are available here: https://github.com/swakkhar/RNA-Editing/.
Any sample in the dataset is a sequence of RNAs. These are all 51 nucleotide long RNA strings. All of these strings are formed by taking symbols from the RNA alphabet, Σ = {A, C, G, U}. Formally a RNA sequence string R ∈ can be formulated as below:
Here, Ni is a nucleotide symbol and L = 51 is the length of the RNA sequence. We have extracted three groups of features from each sequence. Thus each sample in the dataset corresponds to a feature vector containing three groups of features, as below:
The three groups of features that were considered in the PRESa2i method are: k-mer compositions, gapped k-mer compositions and other statistical features. The first two types of features are widely used in the literature for solving other problems as well.
1. k-mer Compositions: k-mers are sequences of length k drawn from the alphabet Σ. Compositions are calculated as the frequency of the different length k-mers normalized by sequence length. This is a widely used feature in the literature15,20. We have used k-mers with value of k = 1, 2, 3, 4. Thus the total number of features in this group was 4+16+64+256=340.
2. Gapped k-mers: We have used gapped k-mer compositions as features. These features were previously used in the literature15,21 and are extended from the ideas of gapped k-mer composition22 or gapped di-peptide composition23. They are normalized frequency of k-mers with gaps in between them. We have considered gaps, g = 1, 2, 3, · · · , 10 in this paper and considered up to k-mers with k = 2, 3, 4. Here, the total number of features is 160+640+640+2560=4000.
3. Other statistical features: We have used other statistically derived features, ratio of start and end codons and distribution of bases, as features. In total, there were five features from this group. The start codon is AUG and end codons are UGA, UAA and UAG. We have used the ratio of these codons. Also there are four nucleotides or bases: A, U, G, C. We have converted the presence of these bases into a distribution and used that as a feature.
Total number of features generated using the feature extraction technique was 4345. The feature generation technique is simple since they are extracted directly from RNA sequences. After the features are selected, we have used a hybrid feature selection technique. Our feature hybrid feature selection method is a multi-step step method where the first step is a wrapper method followed by consecutive filter methods. In the first phase of feature selection we have used best first search (BFS). It is an incremental feature selection procedure that adds or deletes one feature at a time and finds the optimal set of features. After the first phase the selected features were then sent for a single pass classification done by a logistic function (LF). For each class labels, the features with positive weights were considered and others were filtered out in this phase. In the last step, we used ensemble of random decision trees and added most significant features found by the randomly generated features which were left in the previous step to get the final set of features. Figure 1 shows the steps of the feature selection technique used by the PRESa2i method.
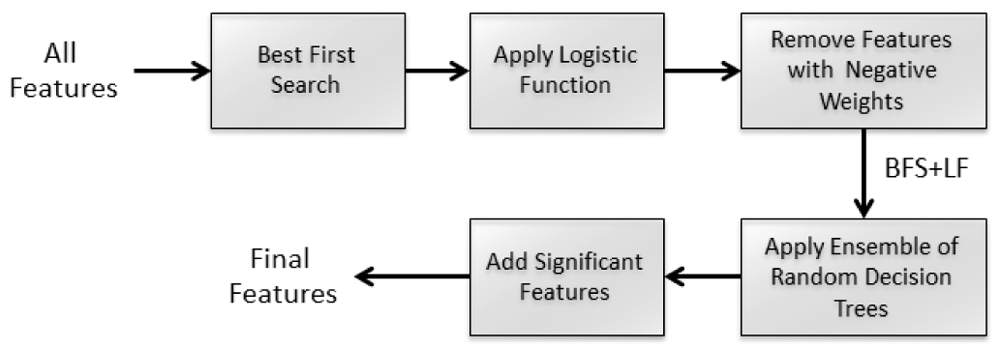
We have used Hoeffding tree24, an incremental decision tree algorithm as the classification engine for PRESa2i. Though its more applicable to data streams, the performance of this classifier is nearly the same as non-incremental learning algorithms. This algorithm starts with a single leaf decision tree and in each iteration for all examples it uses the Hoeffding bound to split the node, recursively selecting one attribute each time based on evaluative functions. The leafs are decided using adaptive naive Bayes technique.
We have used four metrics for the binary classification problem. These are Accuracy, Sensitivity (Sn), Specificity (Sp) and Mathew’s Correlation Coefficient (MCC). These metrics were selected to conform with previous methods8,11. For a binary classification method, let T P denote the number of true positives, T N denote the number of true negatives, F P denote the number of false positives and F N denote the number of false negatives. Now the four metrics are defined as follows:
Note that, the first three metrics have the values in the range [0,1], where a value equal or near 0 means a bad classifier and a value near 1 means a good classifier. However, MCC have values in range [-1,1]. We have also used Receiver Operating Characteristic (ROC) curve to show the performances of different classifiers. It plots true positive rate against false positive rate at different thresholds of a probabilistic classification method.
In this section, we present the details of experimental results for our proposed method, PRESa2i. All the algorithms and programs were developed using Java 8 standard edition and Weka library25. All the experiments were done 10 times and only the average results are reported.
In this section, we provide the experimental results for comparisons with other state-of-the-art methods with our method PRESa2i. We have considered two previous state-of-the-art methods for comparison with our method: PAI proposed by 11 and PAI-SAE proposed by 8. We have used accuracy, sensitivity, specificity and MCC to compare the results. We have not run PAI and PAI-SE, rather as all these methods are tested on the same benchmark dataset, we have taken the results as being reported in the respective publications. Table 2 presents the results on the benchmark dataset. In this table, best values in each criteria are shown in bold fonts.
| Method name | Accuracy | MCC | Sensitivity | Specificity |
|---|---|---|---|---|
| PAI | 79.59% | 0.0600 | 0.8560 | 0. 7311 |
| PAI-SAE | 81.97% | 0.6414 | 0.8720 | 0. 7647 |
| PRESa2i | 86.48% | 0.7393 | 0.9520 | 0.7731 |
From the values reported in Table 2, we can see that PRESa2i achieves higher values in all the performance metrics compared with PAI and PAI-SAE. In case of accuracy our method is 6.89% and 4.51% more accurate than PAI and PAI-SAE, respectively. It also outperforms both of the methods in terms of MCC and sensitivity. In case of specificity its performance is significantly improved compared to other two methods. To further assess the performance of our method we used an independent test set. Note that, while constructing the independent test set CD-HIT was used to remove sequences with higher similarity. We used the same cutoff as being used in PAI proposed by 11. In the case of the independent test set as there were no negative samples, and the only metric applicable was sensitivity. We report the sensitivity of PAI and PRESa2i in Table 3.
| Method Name | PAI26 | PRESa2i |
|---|---|---|
| Sensitivity | 82.33% | 90.67% |
Note that, the sensitivity of our method on the benchmark dataset was not a result of overfit and hence 272 samples out of 300 samples were correctly predicted in the independent test set by PRESa2i compared to 247 correctly predicted by PAI. Thus, we can conclude that our method achieved superior performances in both benchmark dataset and independent test set over these current state-of-the-art methods.
We have proposed a multi-stage novel feature selection method. To show the effectiveness of our feature selection method, we have used accuracy, sensitivity and specificity as performance measures. We have compared the performance of the feature selection method with that of ‘Using All Features’ and with that of ‘Reduced Using BFS+LF’. Results are reported in Table 4. We have used six classifiers: Naive Bayes Classifier, AdaBoost Classifier, Hoeffding Tree, Random Forest Classifier, Support Vector Machines and Bagging.
The bold fonts show the best values of accuracy, sensitivity and specificity for each of the classifiers on the different set of features selected at different stages of feature selection. From this, we can note the effectiveness of the over-all feature selection procedure. Here, we can see that, using best first search and logistic function based feature selection in the first phase generates features that improves the performance of all the classifiers compared to no feature selection. On the other hand, when we further refine the features using decision tree based significance test, all the measures improve for four of the classifiers which is an indication of the effectiveness of the later stage of feature selection.
We can note that among the six classifiers used in our experiments as shown in Table 4, AdaBoost classifier and Hoeffding Tree are the two best classifiers, followed by SVM. In the case of SVM though the accuracy is higher compared to these two algorithms, the sensitivity is not as good as these two. Note that these are the results on the benchmark dataset. We have also shown the receiver operating characteristic curve for each of these classifiers in Figure 2.
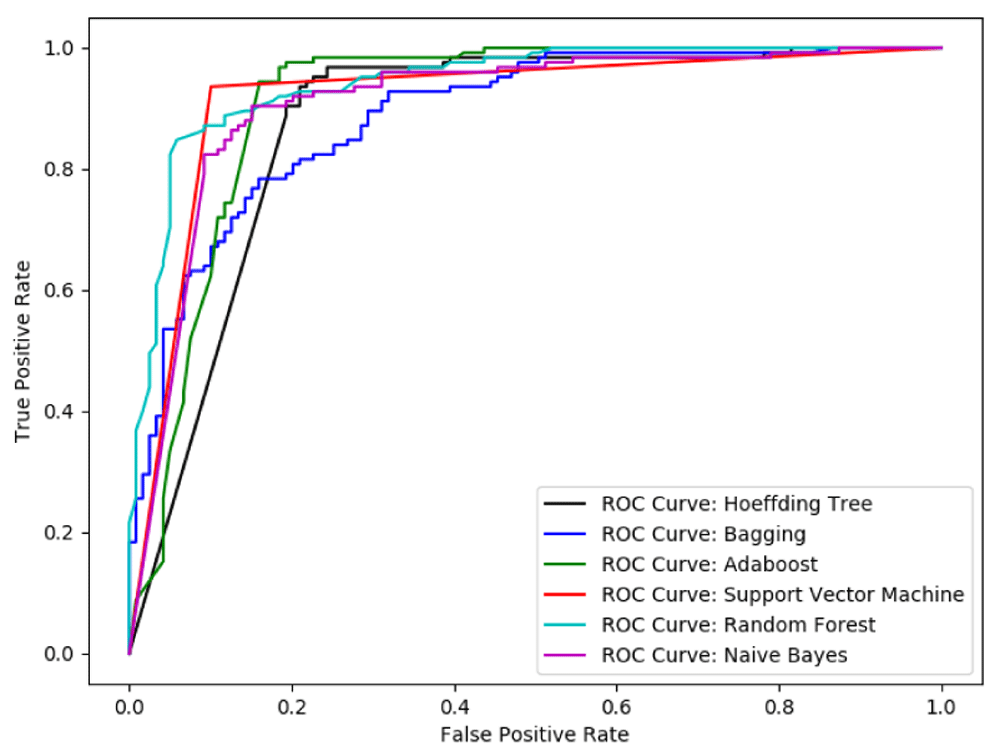
To make the selection of the final classification algorithm, we also tested them on the independent dataset. In Table 5, the results on the independent test set in terms of sensitivity is presented. We see that, the performance of Hoeffding tree does not overfit the data in benchmark dataset and hence the accuracy is enhanced from 86.47% to 90.67% from benchmark dataset to the independent test set. Thus we chose Hoeffding tree as our final classifier and save the model learned by this algorithm for the prediction tool.
We have further investigated the effect of introducing negative instances in the independent test set. For this purpose, we have generated negative instances using a empirical distribution learned from the negative training dataset based on the distribution of the base nucleotides only. The generated negative instance are then added to the positive ones and the algorithms were again tested on this set. The results are reported in Table 6.
| Algorithm | Sn (%) | Sp (%) | Accuracy (%) |
|---|---|---|---|
| Naive Bayes | 86.66 | 73.33 | 80.00 |
| AdaBoost | 85.33 | 51.33 | 68.33 |
| Hoeffding Tree | 90.66 | 52.66 | 71.66 |
| Random Forest | 83.66 | 74.33 | 79.00 |
| SVM | 64.66 | 70.00 | 67.33 |
| Bagging | 82.66 | 61.66 | 72.16 |
From the results reported in Table 6, we note that addition of the negative data in the independent test set does not affect on the effectiveness of the features and the feature selection method and the model trained. Note that, the introduced negative instances are given in the repository at: https://github.com/swakkhar/RNA-Editing/.
Please note that the results presented in Table 6 shows the effect of introducing negative examples in the dataset. Note that these negative datasets are taken randomly and are not verified at all. We note that though the accuracy and sensitivity is still satisfactory, Hoeffding tree and AdaBoost fails in terms of specificity. However, it also indicates that there is scope to improve the performance by adding more negative samples in the training set and also to improve the independent test set by adding real negative examples instead of taking random samples.
We have also implemented a web server, based on the best classification model learned by the incremental decision tree and final selected set of features on the benchmark dataset. Our website was developed using PHP language on the server side using Java and Weka library to provide the prediction from backend. Our method is readily available for use from: http://brl.uiu.ac.bd/presa2i/ index.php.
The web server has two major components: an web interface and a background server application. A small brief on the implementation is given below:
1. Web Interface: The web interface and its backend is implemented using PHP and boostrap. The PHP script takes the input of the RNA sequence and then calls the prediction server which is a Java program and again receives the output from the prediction server and shows them in the web area.
2. Prediction Server: The prediction server is written in Java language. All the experiments are written in java language. The feature generation and selection part is implemented using Java. A program takes the input of the RNA sequence and generates the necessary features and then uses the Weka package for the classification of indentification engine. After the results are processed they are fed back to the Web interface where the results are shown. Interface between the Web Interface and the Java program is made using secured http connection.
The operation of the webserver is very simple. There are only one text field and two buttons. A screenshot of the developed web application is given in Figure 3. Here is a step by step procedure on thow to use the web application:
1. First one has to put the input RNA sequences in the text area. If there is any mistake, pressing the ‘Clear’ button will clear the area.
2. After the input is given in the text area, the predict button should be pressed. Thus a request would be made to the online server.
3. The results for the query sequence will be shown just below the input pane. For the next entry, the process should be followed in the same manner starting from step 1.
In this article, we have proposed PRESa2i, a novel predictor for A-to-I RNA editing sites using sequence based features and incremental decision trees. Our method is based on a number of effective features selected using successive phases of best first search in incremental forward selection and ranking based on random decision trees and logistic regression. We have made all our datasets available online at: https://github.com/swakkhar/RNA-Editing/, and have also made our method available as an web application from: http://brl.uiu.ac.bd/presa2i/index.php. In the future, we would like to explore other RNA editing sites and develop layered or multi-class prediction system. We believe further enhancement of the dataset can improve the performance of the method to a great extent.
Datasets for this article available from: https://github.com/swakkhar/RNA-Editing/
Archived datasets as at time of publication: https://zenodo.org/record/372722127
License: CC0
PRESa2i web application: http://brl.uiu.ac.bd/ presa2i/index.php
Source code available from: https://github.com/swakkhar/RNA-Editing
Archived source code as at time of publication: https://zenodo.org/record/372722127
License: CC0
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)