Keywords
Next-generation sequencing, Variant protein database, NGS-pipeline
This article is included in the Oncology gateway.
Next-generation sequencing, Variant protein database, NGS-pipeline
The new modifications for the articles are as follows
1. We removed some sentences in articles to avoid exceeding the word limit
2. As per the reviewer 1 and 2 comments, we modified the contents in the Introduction and Use case section
3. Supplementary Table 3 has been added to address the reviewer-2 comments
4. References 26-30 has been added
To read any peer review reports and author responses for this article, follow the "read" links in the Open Peer Review table.
Cancer genome sequencing projects have revealed thousands of genomic variations in cancers (Forbes et al., 2010; Tate et al., 2019; Tomczak et al., 2015; Zhang et al., 2011). Some mutants are oncogenic and drive proliferation in various cancers. For example, amino acid substitution of arginine to leucine at position 858 (L858R) in the epidermal growth factor receptor (EGFR) gene has been observed in 17% of pulmonary adenocarcinoma patients (Morgensztern et al., 2015). In addition, a mutation in gene BRAF V600E is known to drive some melanomas (Chapman et al., 2011). Some of these mutant proteins are proteolytically processed in cancer cells, resulting in major histocompatibility complex (MHC) presentation of mutant peptides. These mutant peptides serve as neo-antigens that recruit T cells and result in immune activation (Kreiter et al., 2015). However, not all mutants are encoded at the protein level. Therefore, it is important to identify mutant proteins expressed by cancer cells. However, there are no easy-to-use pipelines for biologists to identify such coding variants, which alter the protein sequences and may play an important role in tumorigenesis. Detection of cancer-specific proteoforms has been studied by several research teams using proteogenomics methods. This approach integrates proteomics data with genome sequence data to identify protein complement of genomic variants (Menschaert & Fenyö, 2017; Nesvizhskii, 2014; Ruggles & Fenyö, 2016). Detection of coding variants can provide candidate molecules for novel therapeutic interventions (Subbannayya et al., 2016).
Several tools have been developed in the last decade to carry out onco-proteogenomics. CPTAC (Ellis et al., 2013) program was initiated to understand the complexity of cancer and its sub types using multi omics approach. Various tools and approaches have been employed to identify mutant peptides in cancers (Mathivanan et al., 2012) (Yeom et al., 2016) (Nagaraj et al., 2015). Several qualitative and quantitative proteomics studies have been reported, which identify altered expression of proteins in cancers (Subbannayya et al., 2015). Often, conventional workflows were used in such investigations, wherein a reference protein database was used to search tandem mass spectrometry data for identification and quantification of proteins (Kelkar et al., 2014). However, such a reference database is usually devoid of sample-specific amino acid variations resulting from genomic alterations. The publicly available databases such as dbSNP (Sherry et al., 2001), COSMIC (Forbes et al., 2015) and UniProt (Apweiler et al., 2004) can be used to identify the variant peptides (Alfaro et al., 2017) but millions of protein-variants from these databases might increase the probability of identifying false positives. Therefore, there is a need for an improved method to identify sample specific sequence variations at the proteomic level. Tools such as CustomProDB (Wang & Zhang, 2013) and MZVar (https://bitbucket.org/sib-pig/mzvar-public/src/master/) have been developed for generating variant protein database. These tools require processed files such as VCF or BED file as an input. Executing the preprocessing steps requires knowledge of computation and also requires multiple tools to generate the output. Hence, we developed CusVarDB with an in-built pipeline for genomics suite to identify variants and create custom variant protein database.
CusVarDB is available at https://sourceforge.net/projects/cusvardb/ and http://bioinfo-tools.com/Downloads/CusVarDB/V1.0.0/. Our tool supports graphical user interface (GUI) for easy execution of next-generation sequencing (NGS) pipelines. The GUI was developed using Microsoft Visual Studio Community edition 2017. Linux commands were executed through the Python program (terminal.py) developed by Linwei available on GitHub. Portable version of Perl was used to execute the Annovar scripts. A python script was used to generate the custom variant database; the scripts were made portable using PyInstaller. The dry-run concept is implemented in the tool to customize commands according to user’s need and run in batches.
CusVarDB inherits different NGS pipelines for genomics, RNA-Seq and exome-seq datasets. The tool performs the following steps: i) alignment of genomic data to a reference genome; ii) variant calling; iii) variant annotation; and iv) generate variant protein database (Figure 1). Burrows-Wheeler Aligner (BWA) is executed for alignment of genome and exome (Li & Durbin, 2009) data, while HISAT2 is used for RNA-Seq data (Kim et al., 2015). Subsequent steps involving sample labelling, variant calling and annotation are performed using Picard, GATK (McKenna et al., 2010) and ANNOVAR (Wang et al., 2010), respectively. CusVarDB substitutes the amino acid variations from the previous steps to generate a custom variant protein database.
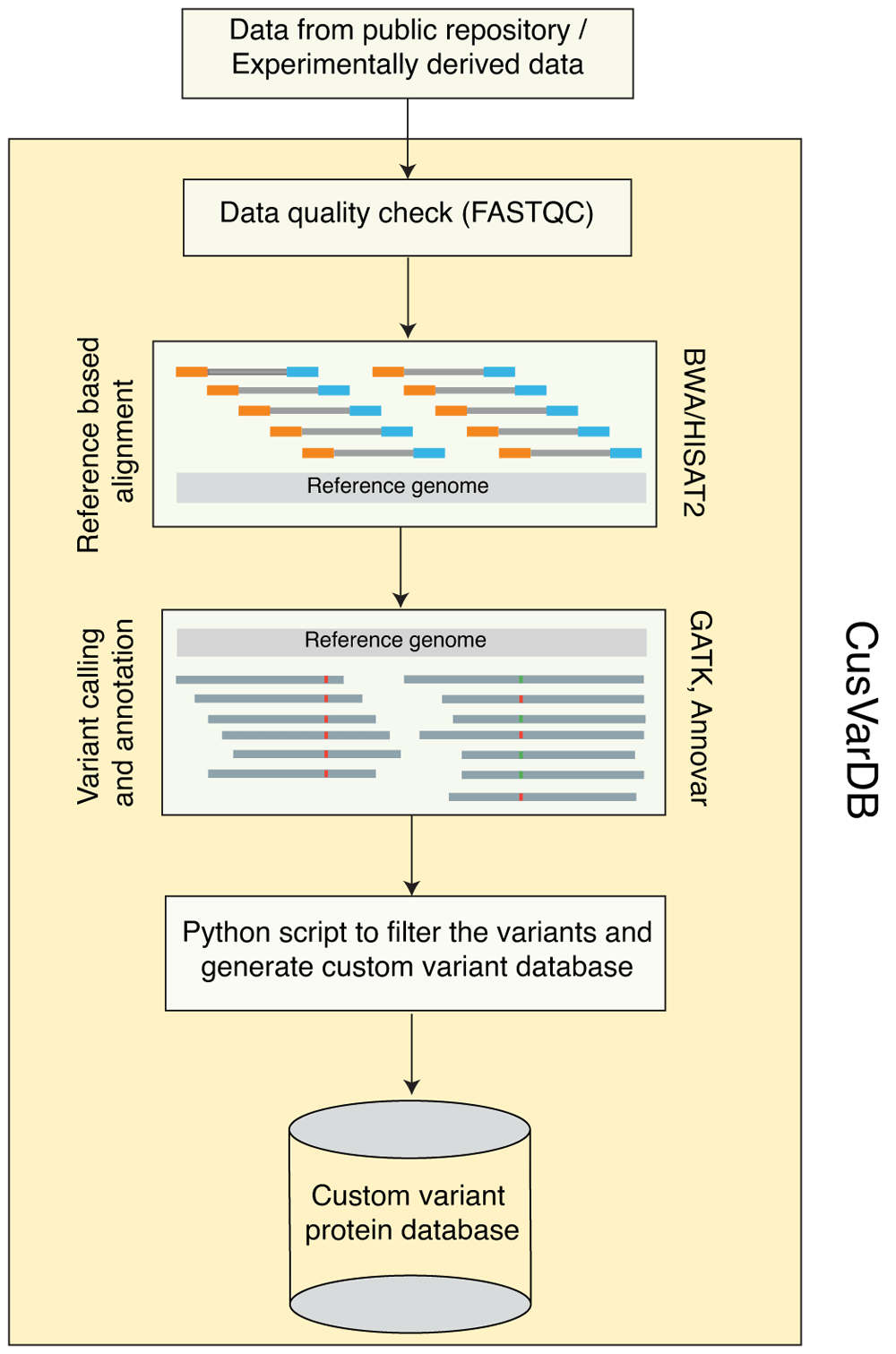
CusVarDB runs on Windows 10 system. It requires Linux Bash Shell and ANNOVAR tool to be enabled and installed by the user. Minimum system requirements include Intel i5 or i7, having at least four cores with 8 GB of RAM and 1 TB hard drive. High performance processors such as Intel i9 or Xeon and large quantity of RAM can enable faster execution of tasks.
The tool requires FASTA, FASTQ and dbSNP database as input files. FASTA file will be loaded at configuration panel and indexed using bwa index or hisat2-build command. The quality control panel loads the FASTQ file and produces a quality check report using the FastQC tool. The third panel asks the user to load the FASTQ file(s), set the number of threads and RAM to perform the alignment and variant calling steps. This step produces the raw VCF file, which will be used for annotation in annotation tab. The annotation panel annotates the variants and incorporates those variants to the protein database to create a custom variant protein database. Detailed information of tool installation and execution of a test dataset are available in the manual (Manual.pdf) available to download here and on Zenodo (Kasaragod et al., 2020b).
The genomics datasets downloaded from the SRA repository were subjected to a quality check using FastQC tool. Poor quality reads with Phred score less than twenty were trimmed using fastx_trimmer. Trimmed reads were mapped to human reference genome version 19 (hg19). AddOrReplaceReadGroup and Markduplicates operations from GATK were performed to add label information and to remove polymerase chain reaction (PCR) artefacts. Further, post alignment processing steps such as IndelRealigner and BaseRecalibrator were performed to correct the mapping errors made by alignment tool to increase variant calling accuracy. Variant calling was performed using HaplotypeCaller. Finally, raw variants were annotated using ANNOVAR and the annotated variants were incorporated to the protein database to create custom variant protein database.
Proteomic searches were carried out using Proteome Discoverer 2.3 (Thermo Scientific, Bremen, Germany). Searches could also be carried out using freely available open source alternatives such as MaxQuant, MsFragger, MS-GF+, MyriMatch or OMSSA. Mass spectrometry raw files were obtained from the PRIDE archive (PXD008222) and searched against the customized variant protein database using SEQUEST-HT search algorithm. Trypsin and LysC were set as proteolytic enzymes with a maximum missed cleavage of one. Carbamidomethylation of cysteine was set as a fixed modification, and acetylation of protein N-terminus and oxidation of methionine were set as variable modifications with a minimum peptide length of seven amino acids. The precursor mass tolerance was fixed as 10 ppm, and 0.05 Da for fragment ions. Mass spectrometry data were searched against the decoy database with 1% false discovery rate cut-off at the peptide level.
We have provided a dataset for users to test the tool. The test dataset was taken from the study SRR7418758 archived on the NCBI Sequence Read Repository (SRA) and aligned using human reference genome version 19 (hg19). Using samtools view command, reads mapped to chromosome 22 were extracted and converted to FASTQ files (paired-end). We have also provided chromosome 22 nucleotide sequences from hg19 and corresponding variant information from dbSNP database.
As a test case, we analyzed exome (Daemen et al., 2013) and proteome datasets (Lawrence et al., 2015) from breast cancer cell lines. We incorporated 12,429; 13,923; 12,386 and 11,600 non-synonymous SNPs from BT474 (accession number SRR925752), MDMAB157 (accession number SRR925788), MFM223 (accession number SRR925796) and HCC38 (accession number SRR925778) cells, respectively, to the protein database (Figure 2A). These non-synonymous SNPs were incorporated to the reference protein database (Human RefSeq release 93) to create customized variant protein database. Mass spectrometry-based raw files were searched against their respective custom variant protein database, which resulted in identification of 423, 408, 386 and 361 variant peptides from BT474, MDMAB157, MFM223 and HCC38, cell line datasets (Figure 2B). Interestingly, we observed mutant protein expression of Replication Timing Regulatory Factor 1 (RIF1) and Torsin-1A-interacting protein 1 (TOR1AIP1) across all four breast cancer cell lines. Mutant plectin (PLEC), marker Of Proliferation Ki-67 (MKI67), HEAT Repeat Containing 1 (HEATR1) and AHNAK nucleoprotein (AHNAK) were detected in three of the four breast cancer cell lines. These coding mutations have also been reported in other cancers The resultant variant peptide lists are available as Underlying data (Kasaragod et al., 2020a). The data generated from the tool demonstrates the usefulness and the ease of detection of variant peptides in an integrated omics analysis.
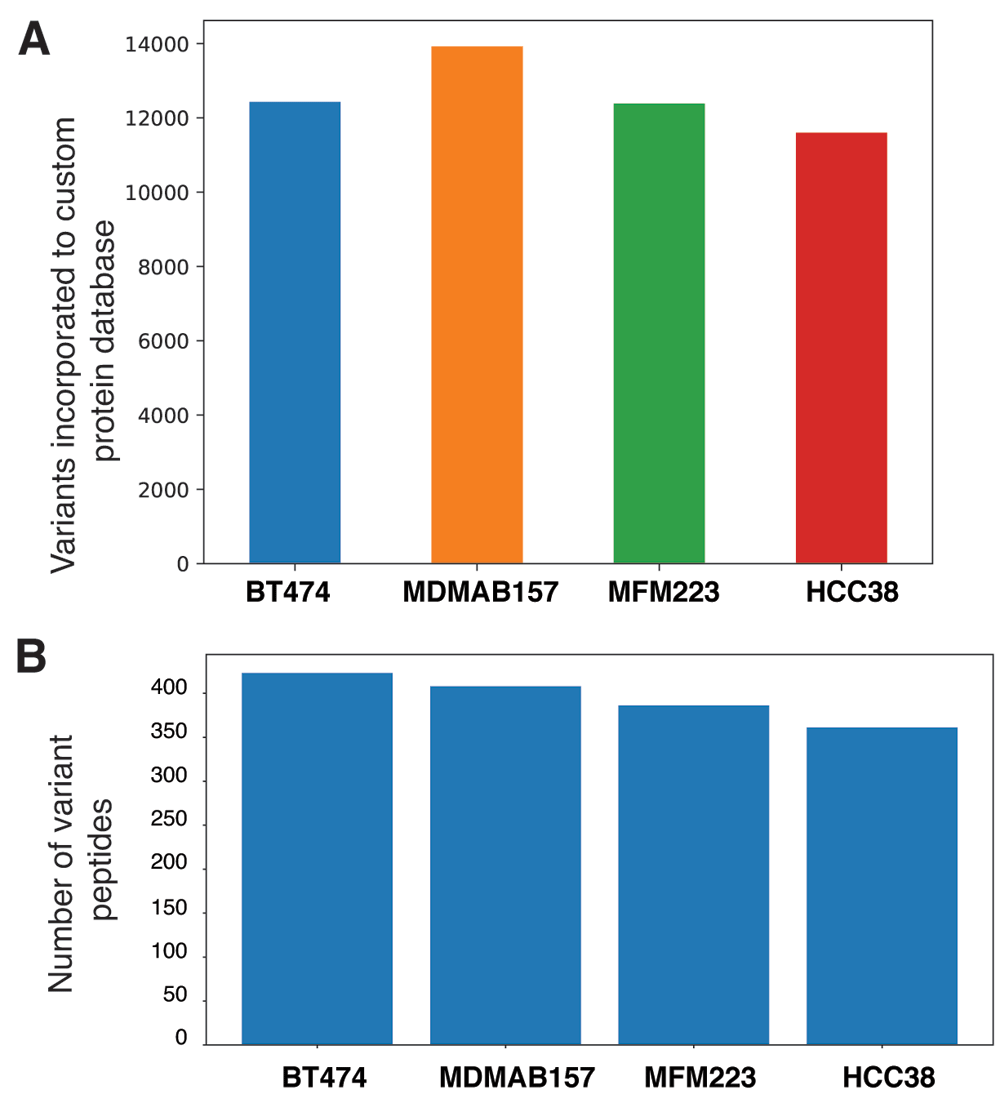
A) Number of nonsynonymous variants incorporated to protein database. B) Variant peptides identified across four cell lines (BT474, MDMAB157, MFM223 and HCC38) by searching mass spectrometry data against sample specific protein database.
CusVarDB generates customized variant protein database from NGS datasets. To our knowledge, this is the first tool which uses Linux Bash Shell to execute the NGS tools on a Windows OS. The tool provides additional feature of dry-run, making the commands highly customizable. The variant protein database generated by this tool is highly compatible for use as a reference protein database for analysis of variants at the proteomic level. CusVarDB currently allows incorporation of non-synonymous mutations. It does not allow incorporation of protein variations resulting from indels and frame-shifts. We developed a flexible tool with the intension of updating it in the future.
Whole exome data from Daemen et al. (2013) on BioProject, Accession number PRJNA210427
Mass spectrometry proteome data from Lawrence et al. (2015) on PRIDE, Accession number PXD008222: https://identifiers.org/pride.project:PXD008222
Test dataset on SRA, Accession number SRR7418758: https://identifiers.org/insdc.sra:SRR7418758
Zenodo: CusVarDB: A tool for building customized sample-specific variant protein database from Next-generation sequencing datasets. http://doi.org/10.5281/zenodo.4018694 (Kasaragod et al., 2020a)
This project contains the following underlying data:
- Gowda_CusVarDB_Supplementary_table1.xlsx (This table contains the resultant variant peptides along with the wild-type peptides from BT474, MDMAB157, MFM223, and HCC38 datasets. Along with mutant peptides, this section also provides additional information such as peptide-spectrum match [PSM], Protein accession, cross-correlation value from the search [Xcorr] and retention time [RT])
- Gowda_CusVarDB_Supplementary_table2.xlsx (This table provides the complete details of the resultant peptides. Here the mutant and corresponding wild-type peptides are mentioned in different sheets. For a given mutant peptide its wild-type peptide and corresponding information can be mapped using the VLOOKUP function in Excel by keeping column A [Sl.No] as lookup parameter.)
- Gowda_CusVarDB_Supplementary_table3.xlsx (This table briefs about the variants which are already reported in other cancers.)
Data are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
Software available from: https://sourceforge.net/projects/cusvardb/ and http://bioinfo-tools.com/Downloads/CusVarDB/V1.0.0/
Source code available from: https://github.com/sandeepkasaragod/CusVarDB
Archived source code at time of publication: https://doi.org/10.5281/zenodo.3780645 (Kasaragod et al., 2020b)
License: Creative Commons Attribution 4.0 International license (CC-BY 4.0).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)