Keywords
Hurler syndrome, mucopolysaccharide, enzyme, genetic
Hurler syndrome, mucopolysaccharide, enzyme, genetic
Mucopolysaccharidosis (MPS) represent a set of metabolic disorders, which are autosomal inherited disorders. MPS are lysosomal storage disorders that occur as a result of the deficiency of one group of enzymes, which degrade three classes of mucopolysaccharides: heparan sulphate, dermatan sulfate, and keratan sulfate1. MPS occurs due to mutations in the gene encoding human α-L-iduronidase2. Glycosaminoglycans (GAGs) accumulate chronically and progressively in the lysosomes of the cells throughout the body. Such accumulation of GAGs leads to multiorgan dysfunction and significant morbidity. Patients with Hurler’s syndrome (MPS Type 1) experience progressive debilitation of the musculoskeletal, cardiorespiratory, and central nervous systems, leading to death before 10 years of age if they remain untreated. There are very few cases of Hurler syndrome reported in Pakistan especially in last few years. Here, we present the case of Hurler’s syndrome in a young female patient.
An 8-year-old female patient was referred to the Children’s Hospital, Faisalabad, Pakistan with complaints of respiratory distress, massive abdominal distension, and generalized body swelling. According to her parents, developmental milestones slowed down at the age of 1.5 years with gradual mental decline expressed in terms of lost physical skills. The parents mentioned multiple joint stiffnesses and that the patient was having difficulty in walking for last few years and was unable to walk in the last year of her life. Though they were unaware of any hearing difficulties in the patient, they reported that she was not able to speak. The patient had noisy breathing noticeable since the third day of birth. In the past few years, the patient had multiple chest infections and had been constipated for around the last 3–4 months before hospital admission. Family history revealed that the parents had a consanguineous marriage (first cousins), and the patient had one older brother who had no obvious mental and physical abnormalities. This was the patient’s first tertiary hospital visit.
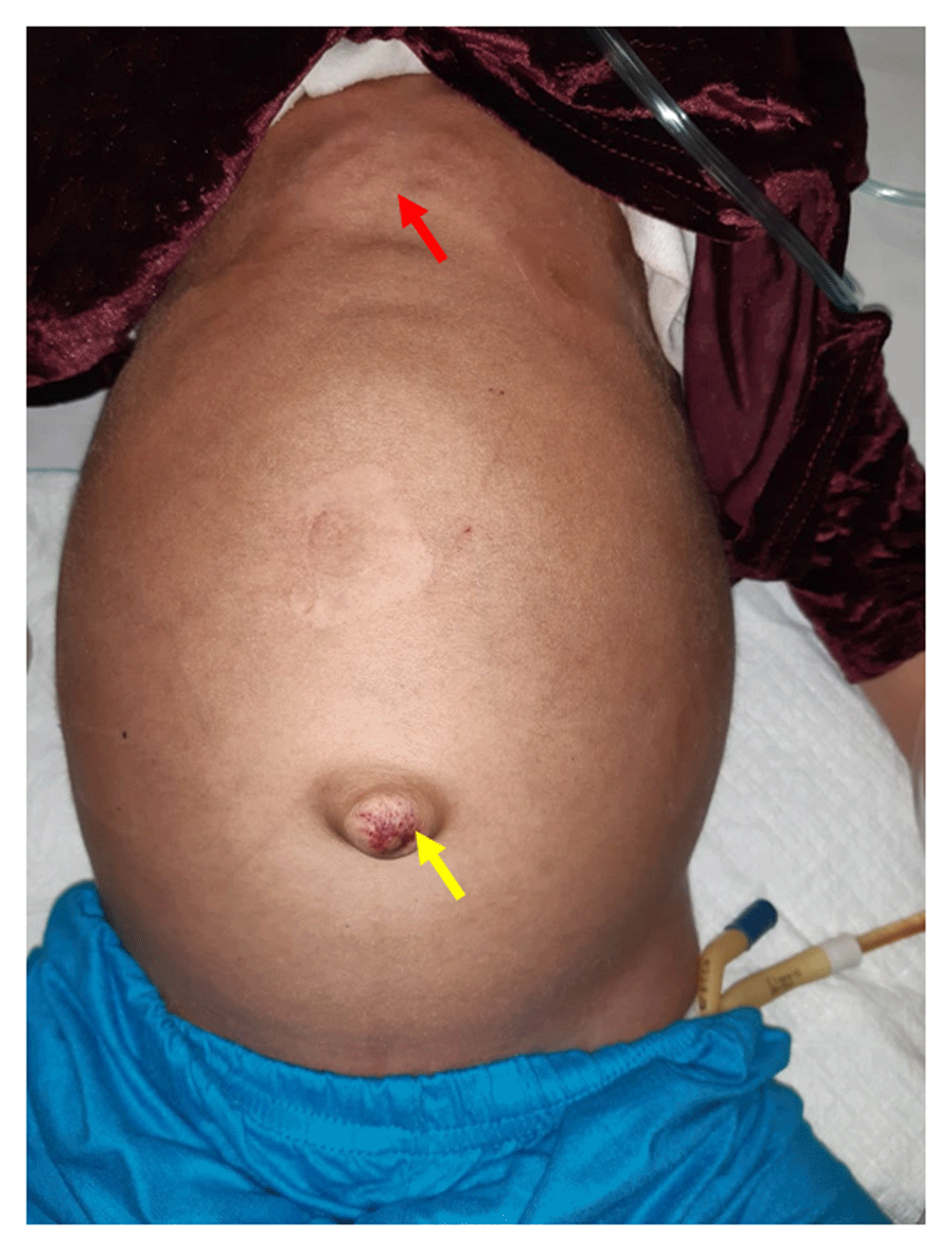
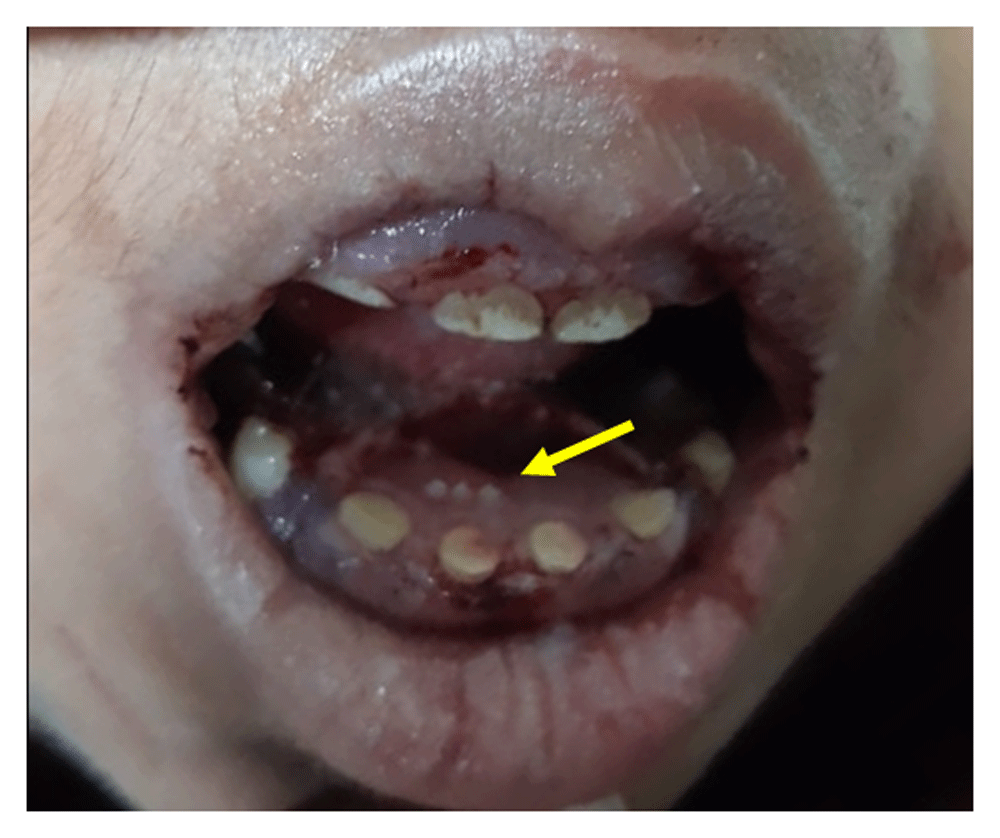
General physical examination revealed coarse facial features, hirsutism, low set ears, depressed nasal bridge, rotated legs, talipes varus, short neck, distended abdomen, pectus carinatum, thick short claw-like hands, umbilical hernia, and kyphosis (Figure 1). Abdominal examination revealed massive hepatosplenomegaly. On cardiac auscultation, a murmur was not heard but loud P2 was audible. Moreover, on chest auscultation, bilateral crepitations were present. On oral examination, gingival hypertrophy, spaced dentition, misaligned eruptive permanent dentition, microdontia, delayed eruption of the permanent tooth and delayed shedding of the deciduous tooth were observed (Figure 2).
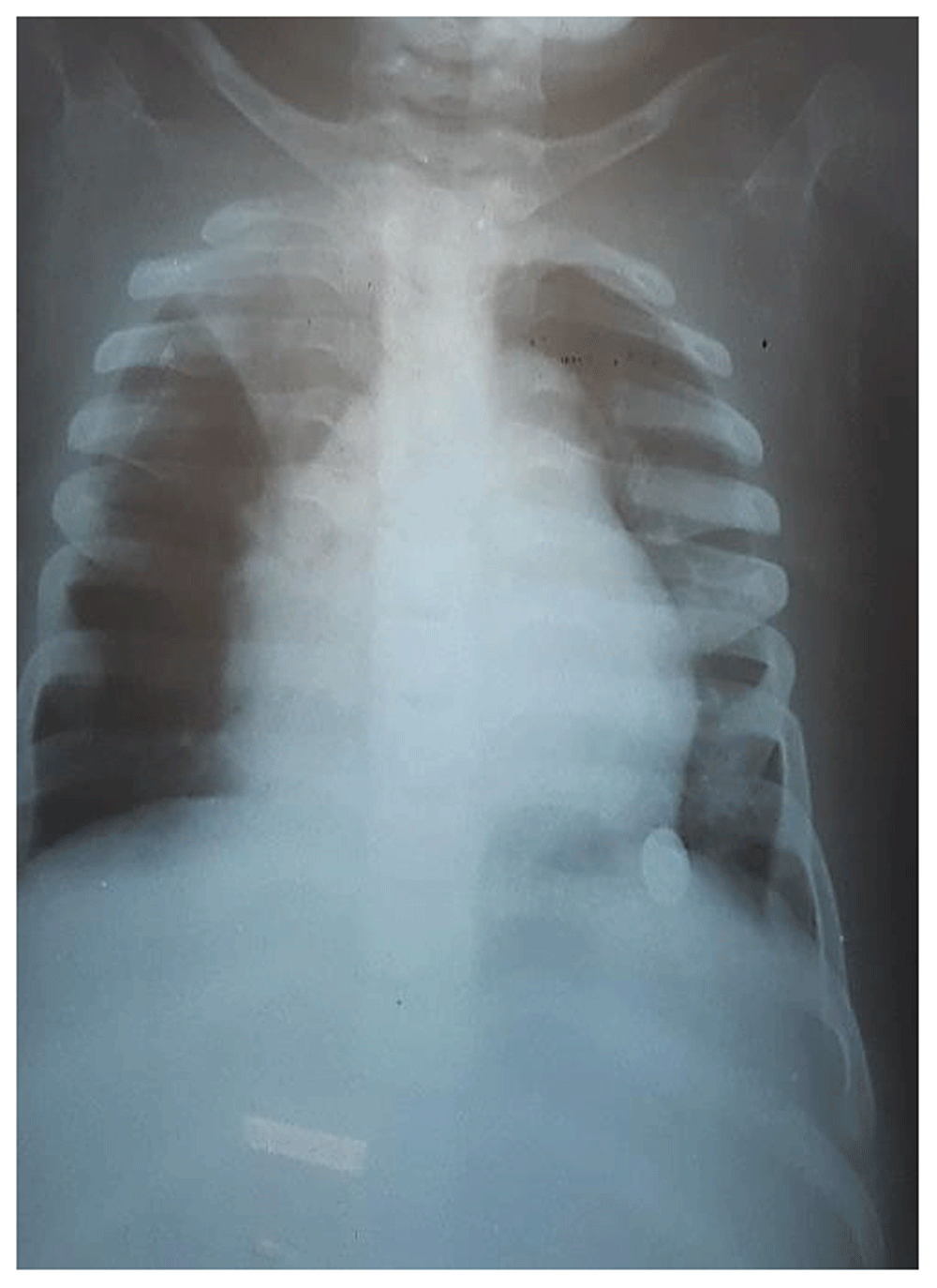
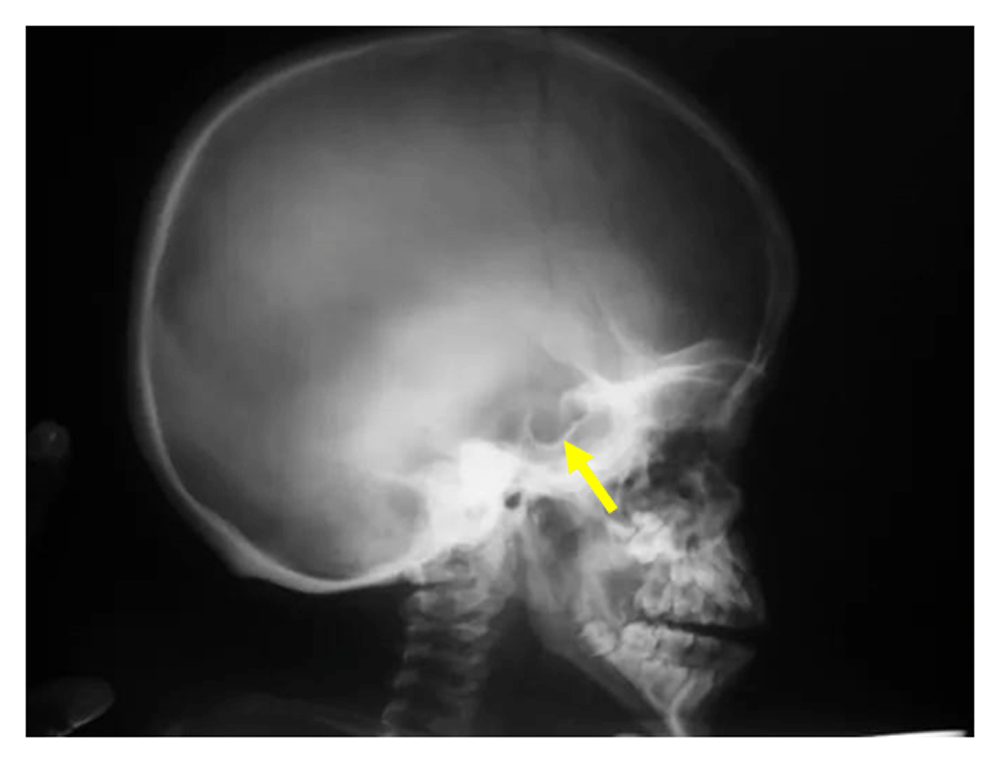
Chest X-ray showed cardiomegaly and oar shaped ribs (Figure 3). X-ray of the patient’s skull showed J-shaped Sella Turcica (Figure 4), while X-ray of her hand showed proximal pointing of metacarpals (Figure 5), and X-ray of the lumbosacral spine showed inferior beaking of vertebrae (Figure 6). The patient’s echocardiogram showed normal left ventricular function, and hematological investigations were all in the normal range.
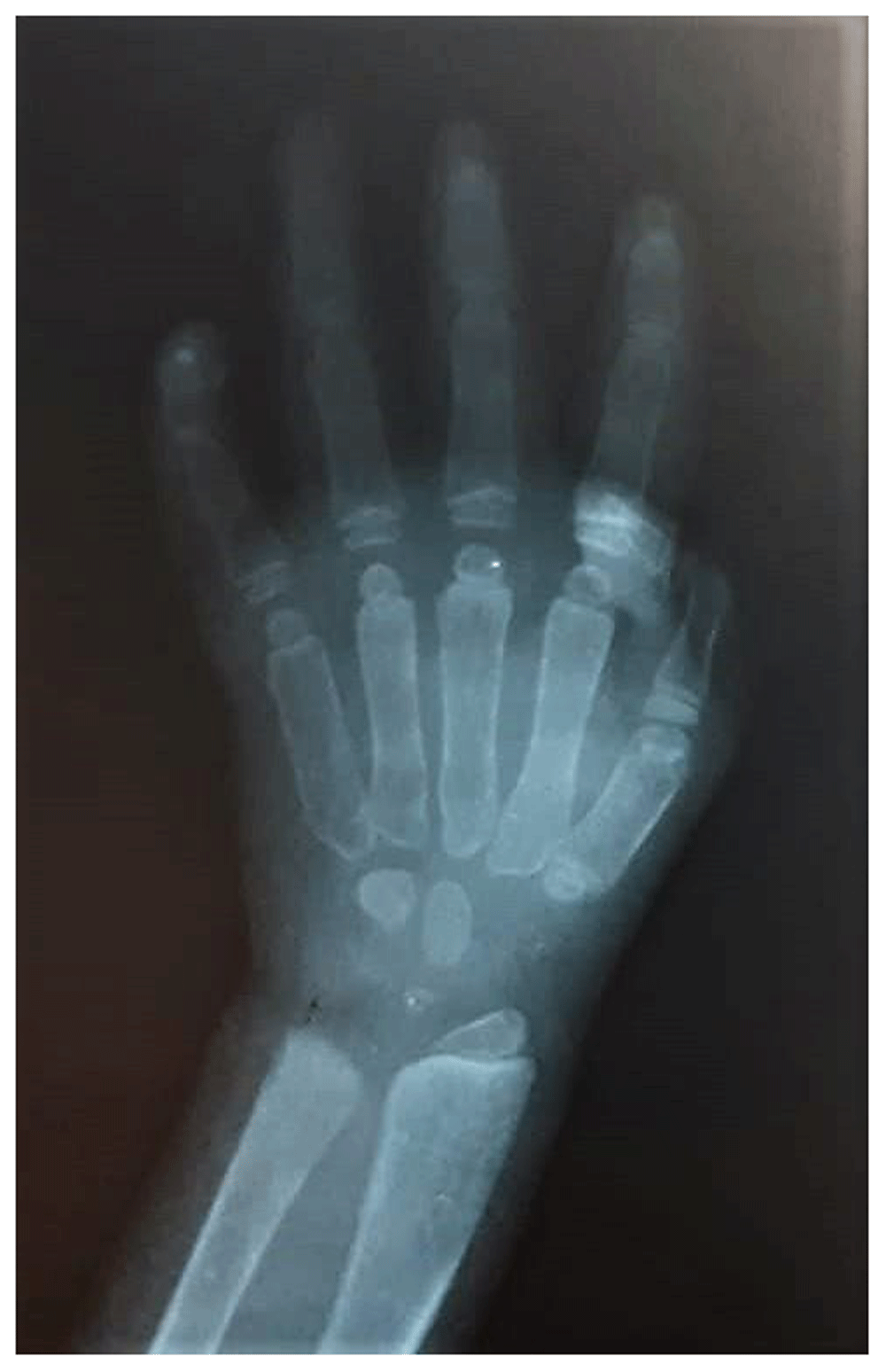
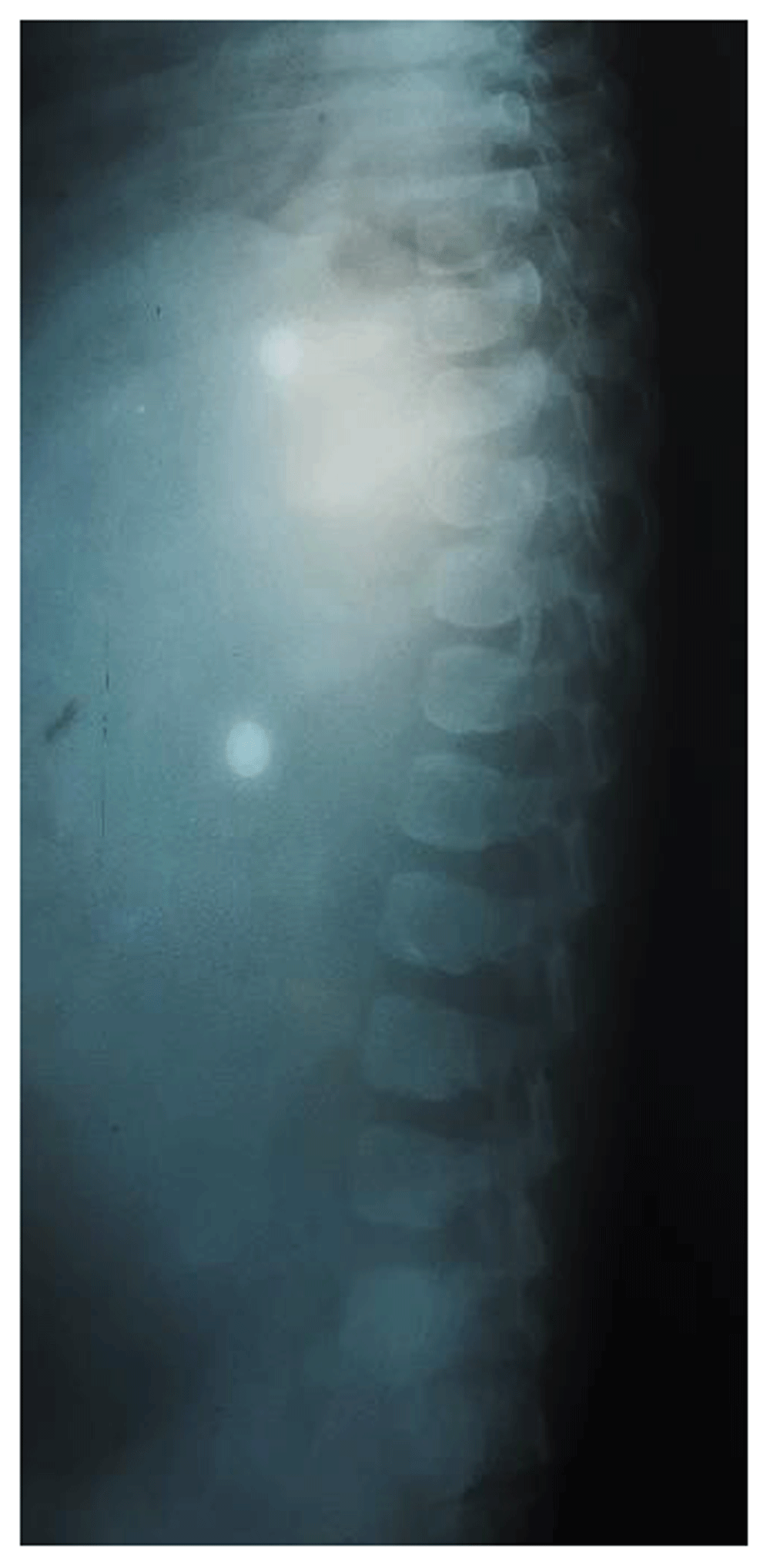
A differential diagnosis of Hunter syndrome (MPS Type II; MPS-II) and Sly syndrome (MPS-VII) was made. However, after clinical analysis and imaging findings, a provisional diagnosis of Hurler syndrome was made. The patient’s MPS urine was 1673 mL MPS/g creatinine (normal levels, 116.4–324.4 mL MPS/g creatinine). The patient’s α-L-iduronidase activity in the leukocytes was non-detectable (normal levels, 0.105–0.327µmol phenol per 18 hours per mg protein). A final diagnosis of Hurler syndrome (MPS-I) was confirmed after the result of urinary excretion of MPS and an enzyme assay.
On arrival the patient was having difficulty breathing. She was supplied with oxygen through nasal prongs and was managed conservatively. On the third day, the patient was intubated and kept on ventilatory support. On diagnosis of Hurler syndrome (MPS-I), the patient’s parents were advised for further management; however, they denied further management because of socio-economic reasons. The patient died on her fifth day of hospital stay.
Hurler syndrome manifests as an autosomal recessive disorder of mucopolysaccharide metabolism. There is an excess accumulation of lipids in the central nervous system as well as other viscera. This condition manifests in early infancy. Patients have developmental retardation, an expressionless face, and increase in the size of the head with deformed shape. They also have various skeletal abnormalities, contracture on flexion, hernias, and an increase in the size of the liver and spleen. Corneal clouding can be found in some patients. Hurler syndrome is a metabolic disorder of mucopolysaccharide metabolism to defect in lysosomal degradation pathways. It is seen in approximately 1:100,000 live births3.
Hurler syndrome is diagnosed based on the reduced level of α-L-iduronidase activity in leukocytes or cultured fibroblasts4. Prenatal diagnosis is confirmed by the presence of unusual glycosaminoglycan components in the amniotic fluid, or abnormal metabolic activity in cultures amniotic fluid cells, and a deficiency of the lysosomal enzyme α-L-iduronidase in these cell homogenates5. Along with symptomatic treatment, enzyme replacement therapy with α-L-iduronidase, as well as bone marrow transplantation, increases the probability of life expectancy6. Couples who have a positive family history must be provided with genetic counseling and testing.
Hurler syndrome is a rare genetic disorder of mucopolysaccharide metabolism and is caused by a defect in lysosomal degradation pathways. For patients with MPS-I, enzyme replacement modality of treatment can increase the patient's survival rate if an early diagnosis can be made.
Written informed consent was obtained from the father of the patient for the publication of this case report and associated images.
All data underlying the results are available as part of the article and no additional source data are required.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)