Keywords
Intervening sequence, IVS1nt5, IVS1nt1, beta globin gene, RNA structure, beta thalassemia major
Intervening sequence, IVS1nt5, IVS1nt1, beta globin gene, RNA structure, beta thalassemia major
We modified the view of figure 1. Only the mutations area was displayed in previously published, whereas complete paired sequences are retrieved in this version.
See the authors' detailed response to the review by Kholis Abdurachim Audah
See the authors' detailed response to the review by Zhichao Miao
Thalassemia is a hereditary blood disorder that induces the production of an abnormal form or inadequate amount of hemoglobin in the body. Hemoglobinopathy has been identified in approximately 71% of all countries around the world, and more than 50% of cases of β-thalassemia occur in Southeast Asia1. β-thalassemia is one of the most prevalent blood disorders worldwide. It can result in various traits and the coinheritance of thalassemia minor, intermedia, or major depends on many factors. People with β-thalassemia exhibit reduced hemoglobin production, and low levels of hemoglobin results in a lack of oxygen supply throughout the body2.
β-thalassemia is caused by mutations in the human beta-globin (HBB) gene, which is responsible for producing β-globin, a subunit of hemoglobin3. Most mutations associated with β-thalassemia are caused by the substitution of one or a limited number of nucleotides in HBB. These mutations affect the functions of the gene including transcription, RNA processing or translation of β-globin mRNA. β zero (β0)-thalassemia is caused by mutations in HBB that stop the production of beta-globin. Conversely, other mutations only reduce the amount of beta-globin protein produced, a condition termed β plus (β+)-thalassemia4.
People with β0- and β+-thalassemia have been diagnosed with thalassemia major and thalassemia intermedia, respectively. Thalassemia major has a more severe phenotype than thalassemia intermedia. Most patients with thalassemia major die at a young age5. Unfortunately, as many as 23,239 babies are born with inherited β-thalassemia major every year1.
Conformational changes in regulatory RNA induce specific human diseases. More than 95% of mutations result in only small local changes in the conformation of RNA. The same phenotype (disease) can be caused by different mutations with varying degrees of effects on the overall RNA structure6.
Numerous variants of non-deletional β-thalassemia are caused by mutations that interfere with processing of the primary mRNA transcript7. These mutations affect AG or GT dinucleotides at the exon-intron splice junction. These mutations eventually ablate regular splicing and induce β0-thalassemia, resulting in a β-thalassemia major phenotype4. Mutations in intervening sequence 1 (IVS1) at nucleotides 143 (change from guanine to thymine, IVS1nt1G>T) and 147 (change from guanine to cytosine, IVS1nt5G>C) are common mutations in Southeast Asia that result in β-thalassemia minor and major, respectively8.
A study on mutations that interfere with mRNA transcription may explain the mechanism by which the mRNA structure is changed by mutations in HBB. Specific in silico RNA analysis and visualization tools to study these structural changes have been developed, such as ViennaRNA Web Services, 3dRNA v2.0, and UCSF Chimera9–12. ViennaRNA Web Services and 3dRNA v2.0 are web interface packages that offer structure modeling for RNA. This study evaluated the effects of the intronic HBB mutations IVS1nt1 and IVS1nt5 on the RNA structure using specific in silico tools.
The complete genome sequence of HBB was obtained from Ensembl for transcriptional regulation detail by selecting transcript HBB-201 ENST00000335295.413. Concerning mutations in IVS1, introns 1–2 were mutated manually at nucleotides 143 (IVS1nt1G>T) and 147 (IVS1nt5G>C). The sequences of wild-type and mutant HBB were aligned using ClustalW Multiple alignment in BioEdit version 7.214. The complete wild-type and mutant sequences were transcribed into mRNA using Nucleic Acid Converter (https://skaminsky115.github.io/nac/).
We used the RNAfold Server from ViennaRNA Web Services to predict the RNA secondary structures9. By selecting this applet, thermodynamic parameters, including centroids, partition function, and minimum free energy (MFE), were automatically calculated by the software as considered for structure folding. To create the RNA best structure tree plot, the wild-type and mutants sequences and structural elucidations were forwarded to the Barriers server, which is part of the ViennaRNA Web Services package15,16. The tools were utilized with their respective default parameters.
Free energy minimization and base pairing probabilities are the most used methods in predicting single-sequence structure using the following equations17. When a structure i is at equilibrium with the unpaired state, the equilibrium is described by the equilibrium constant Ki as presented in Equation 1:
The Gibbs free energy change for structure i, namely ΔGi, quantifies the favorability of a given secondary structure compared with the unpaired state. Similarly, the equilibrium between two structures i and j is described in Equation 2 as follows:
Thus, the Gibbs free energy change quantifies the favorability of a structure at a given temperature. The structure with the lowest Gibbs free energy change will be the most prevalent in solution at equilibrium.
Supplementing a free energy minimization calculation with a partition function calculation can identify the pairs that are more likely to be correct. The partition function Q is defined as the sum of the equilibrium constants Ki for all possible structures.
Thus, the probability of a particular structure i being found in solution can be calculated according to Equation 4 as follows:
Furthermore, the probability of a base pair i–j can be calculated by summing the equilibrium constants for structures that contain that pair and dividing by the partition function.
In Equation 5, the sum of the index k is taken over all structures that have the base pair i–j.
The 3dRNA v2.0 from Biomolecular Physics and Modeling Group Xiao Lab (http://biophy.hust.edu.cn/services.html) was used to construct the tertiary structures. Whole genome sequence and 2D structure with dot-bracket form, obtained from RNAfold Server, were entered to generate 3D structures for wild-type and each mutant10. The prediction files were downloaded in the .pdb format. The interactive model was visualized using the University of California at San Francisco (UCSF) Chimera version 1.13.118.
Intronic mutations of HBB can lead to β-thalassemia major. These mutations have spread widely around the world19. In this study, we observed the effects of intronic mutations on HBB mRNA structure. HBB is encoded by 3931 nucleotides located on chromosome 11 with its various regulatory genes. This gene contains 1608 bp and consists of three exons and two introns. Introns 1–2 cover nucleotides 143–272. This region is known as IVS1 (Figure 1a). In these introns, we highlighted two mutations, namely IVS1nt5G>C and IVS1nt1G>T, located at nucleotides 143 and 147, respectively (Figure 1b).
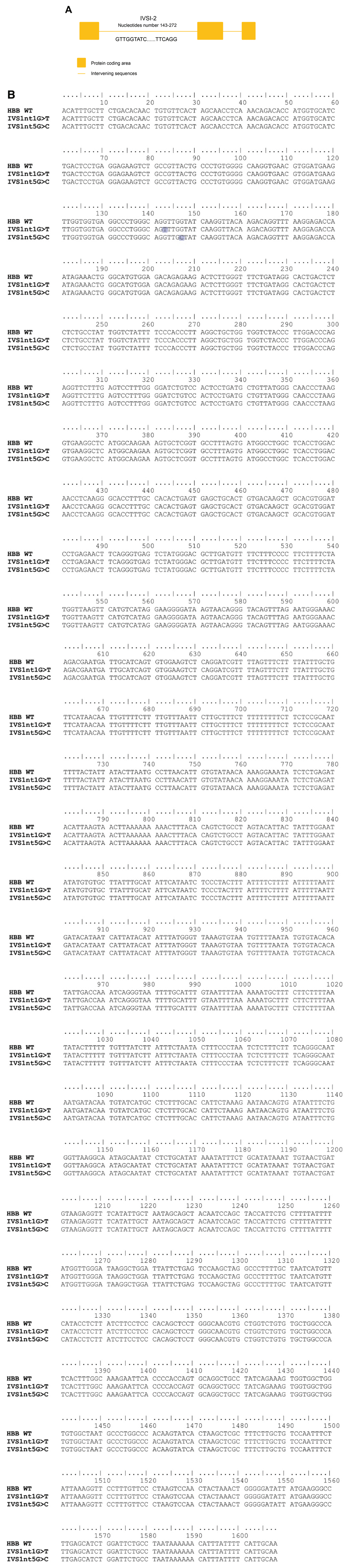
(A) Details of HBB showing the target mutations. (B) Sequence alignment of wild-type and mutant HBB was performed using ClustalW Multiple alignment in BioEdit version 7.2, mutations were highlighted in blue.
Our observation revealed differences in the MFE and other thermodynamic characteristics between wild-type and mutant HBB. We found that different point mutations occurred in the same IVS region, reflecting unique features. MFE, centroid, and partition function properties are depicted in a mountain plot in Figure 2.
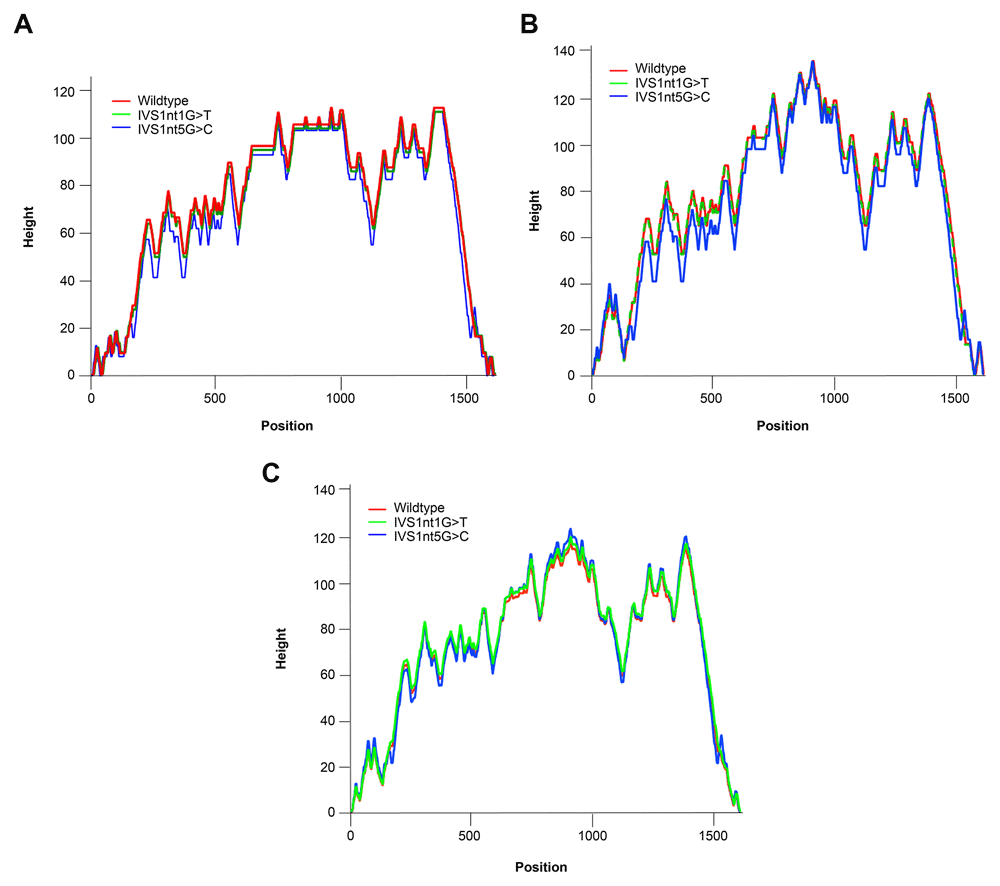
(A) Centroid, (B) minimum free energy, and (C) partition function.
The MFE has important roles for secondary structure prediction. Lower values are associated with better structures. Conformation of MFE secondary structure by RNA folding is irreversible because the sequence space sets up the shape space20. Centroid-based analysis is an alternative method for predicting the secondary structure. This calculation yields 30% less prediction errors than the MFE21. The stability of RNA molecules fluctuates in different low energy structure. Therefore, we need to analyze the equilibrium ensemble of RNA structures via the partition function22. The partition function measures the quality of secondary structure by determining the confidence in base pairs predicted by free energy minimization23.
Based on the mountain plot result, the mutations of IVS1nt1G>T and IVS1nt5G>C changes the mRNA structure of HBB. However, IVS1nt5G>C leads to a much larger free energy change versus the wild-type. A previous study found that IVS1nt1G>T and IVS1nt5G>C take place in splice junction and consensus splice site, respectively, that might result in splice junction disturbances24. Therefore, we believe that this disturbance may affect the mRNA structure.
Although introns are known as non-coding regions, their alteration influences mRNA expression, which can lead to several diseases. Using the RNAfold applet from the ViennaRNA package, we analyzed the thermodynamic characteristics and structural changes of wild-type and mutant HBB (Figure 3 and Figure 4)7,9.
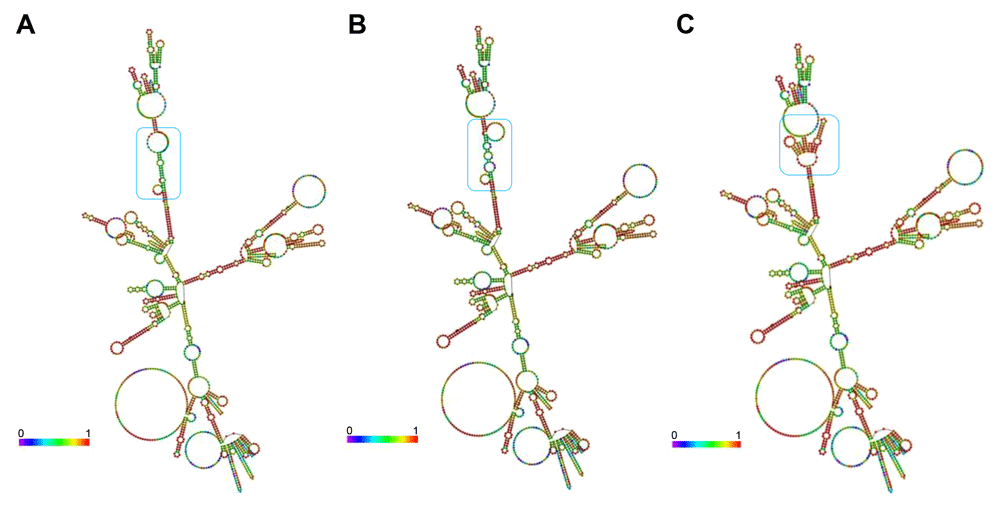
(A) Wild-type, (B) IVS1nt1G>T, and (C) IVS1nt5G>C. Structural changes of RNA are highlighted by blue boxes.
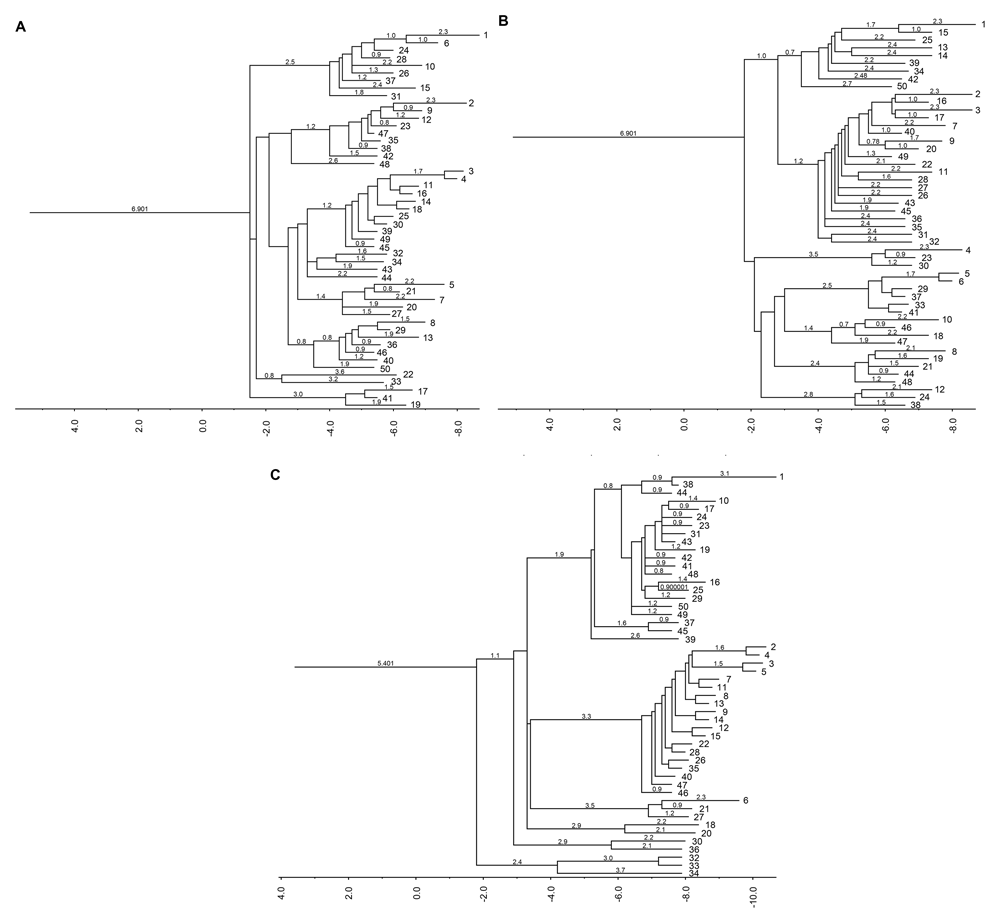
(A) Wild-type, (B) IVS1nt1G>T, and (C) IVS1nt5G>C.
The secondary structure of mRNA is beneficial for tracing the refolding pathways of an RNA molecule. The next question that arises is how the secondary structure of mRNA is affected by mutation of HBB. To obtain the mRNA structure, the RNAfold system was used (see Methods). The prediction of the mRNA secondary structure is based on the centroid-based pair probability (Figure 3). It was found that the three structures have a distinct structural backbone. Based on the structure, unwanted folding occurred in the IVS1nt1G>T and IVS1nt5G>C mRNA structures compared with the wild-type structure. The principle of this methods is to create the secondary structure with minimal base pair distance to all other secondary structures in the Boltzmann ensemble9. The blue box in Figure 3 highlighted the main alteration formed within the three sequences. However, the structure of IVS1nt5G>C seems to be the best predicted according to the red color in the base-pairing probability. To further clarify the effect of folding, barrier tree plot analysis was performed (Figure 4). The result revealed that changes in the population occur earlier in IVS1nt5G>C carriers than in people carrying wild-type or IVS1nt1G>T HBB. The earlier changes in the population result in different populations toward the end of the barrier tree plot.
These intronic HBB mutations may lead to critical folding changes that could drastically affect the tertiary structure of the beta-globin chain as well as the quaternary structure. To further clarify the effects of the mutations, 3D models of wild-type and mutant and HBB were visualized using UCSF Chimera software (Figure 5). In general, the tertiary structure of each sequence was unique. The point mutation gave tremendous folding changes in the 3D model. Once the base shifted, base pairing mechanism was influenced massively. Previous research reported the predicted pathogenic effects of these intronic mutations using the HSF server. IVS1nt5G>C was predicted to be a silencer motif new site, intronic identifying elements and exonic splicing silencer (ESS) site were broken. ESS provides binding sites for proteins, promoting exon exclusion, and helps the spliceosome to decoy splice sites25. In addition, Seo et al. (2013) stated that most of the mutations affecting splicing disrupted the highly conserved donor and acceptor sites (GT/AG) at the exon–intron junctions and polypyrimidine tract, and the branch-point sequence may be disrupted by mutations affecting the splicing sites26.
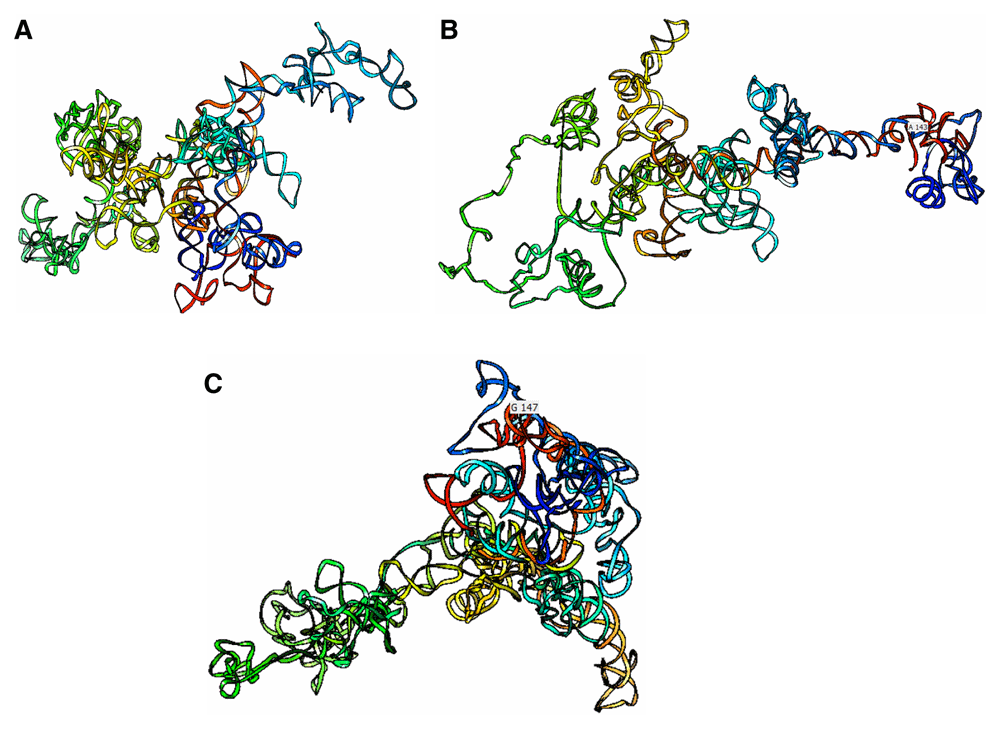
This prediction shows complex structural folding considering the base pairing probability.
RNA structure stability depends on metal or ligand binding, the alteration of which can affect the binding site activity and structure stability. The properties of RNA structures related to multiple ligand binding, ligand binding-induced conformational changes, and the ion-stabilized catalytic core have been explored. The study by Miao et al. stated that the positive charge of metal cations plays compensates for the negative charge of the RNA phosphate backbone27. In addition to that function, metal ions also bind to extremely specific locations on RNA 3D structures27. All of these findings reveal that IVS1nt5G>C significantly changes the mRNA structure. However, the protein structure of HBB still needs to be elucidated to understand the effects of the mutations on protein features after translation.
The mRNA plays important roles in protein regulation. RNAs start to form after the transcription process is completed. Once done, the structure of RNA is critical for its activities. Both IVS1nt5G>C and IVS1nt1G>T give implications to the clinical condition due to protein instability. There are some disruptions in producing beta globin chains by these mutations, hence the beta thalassemia major (BTM) patients produce beta globin chain in a limited number or even not at all. This mechanism results in unbalanced alpha and beta globin chains in hemoglobin that expedite the hemolysis and ineffective erythropoiesis contributing to reduced production of mature red blood cells28. Thus, the BTM patients requires lifelong blood transfusions and commonly suffer complications, such as endocrine complications and cardiac complications29.
This study examined the effects of mutations in terms of the minimum energy and the impact on the secondary and tertiary structures of HBB mRNA. Changes in the RNA formation may lead to disturbances of the protein structure. Protein expression can be influenced by mRNA folding, other local and global sequence features, and modulation of mRNA stability by codon usage30. Regarding the length of introns 1–2, the mutations occur in small RNA-affected regions where analysis is important for detection, annotation, quantification, and de novo discovery of putative small RNA molecules31. However, it will be interesting to clarify the effects of splicing, debranching, and trimming on the mRNA structure in future research.
Our research revealed the impact of intronic mutations on the secondary and tertiary structure of mRNA. Different point mutations occurring in the same IVS region were associated with unique characteristics. We have expanded our understanding of the thermodynamic aspects of these mutants. The limitations of this study included our inability to clarify the interaction of secondary structures due to the lack of an RNA analysis platform function for intronic mutations. RNA molecular simulation and wet lab experiments are required to elucidate the detailed features of the intronic mutants and their interactions with ions or ligands to further assess their functional failure, especially in genetic disease.
Figshare: DNA sequence of wild-type and intronic mutation human beta-globin gene, https://doi.org/10.6084/m9.figshare.11565726.v232.
Figshare: RNA sequence of wild-type and intronic mutation human beta-globin gene, https://doi.org/10.6084/m9.figshare.11566596.v133.
Figshare: Mountain plot of wild-type and intronic mutation human beta-globin gene, https://doi.org/10.6084/m9.figshare.11556675.v334.
Figshare: Centroid base pair probability of wild-type and intronic mutation human beta-globin gene, https://doi.org/10.6084/m9.figshare.11565711.v235.
Data are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)