Keywords
malaria, Plasmodium falciparum, machine learning, parallel computing, Apache Spark, big data, artemisinin, bioinformatics, DREAM Competition
This article is included in the Software and Hardware Engineering gateway.
This article is included in the Artificial Intelligence and Machine Learning gateway.
malaria, Plasmodium falciparum, machine learning, parallel computing, Apache Spark, big data, artemisinin, bioinformatics, DREAM Competition
Malaria is a serious disease caused by parasites belonging to the genus Plasmodium which are transmitted by Anopheles mosquitoes in the genus. The World Health Organization (WHO) reports that there were 219 million cases of malaria in 2017 across 87 countries1. Plasmodium falciparum poses one of greatest health threats in Southeast Asia, being responsible for 62.8% of malaria cases in the region in 20171.
Artemisinin-based therapies are among the best treatment options for malaria caused by P. falciparum2. However, emergence of artemisinin resistance in Thailand and Cambodia in 2007 has been cause for research3. While there are polymorphisms in the kelch domain–carrying protein K13 in P. falciparum that are known to be associated with artemisinin resistance, the underlying molecular mechanism that confers resistance remains unknown4. The established pharmacodynamics benchmark for P. falciparum sensitivity to artemisinin-based therapy is the parasite clearance rate5,6. Resistance to artemisinin-based therapy is considered to be present with a parasite clearance rate greater than five hours7. By understanding the genetic factors that affect resistance in malaria, targeted development can occur in an effort to abate further resistance or infections of resistant strains.
First, we created a machine learning model to predict the IC50 of malaria parasites based on transcription profiles of experimentally-tested isolates. IC50, also known as the half maximal inhibitory concentration, is the drug concentration at which 50% of parasites die. This value indicates a population of parasites’ ability to withstand various doses of anti-malarial drugs, such as artemisinin.
Training data was obtained from the 2019 DREAM Malaria Challenge8,9. The training data consists of gene expression data of 5,540 genes of 30 isolates from the malaria parasite, Plasmodium falciparum. For each malaria parasite isolate, transcription data was collected at two time points [6 hours post invasion (hpi) and 24 hpi], with and without treatment of dihydroartemisinin (the metabolically active form of artemisinin), each with a biological replicate. This yields a total of at eight data points for each isolate. The initial form of the training dataset contains 272 rows and 5,546 columns, as shown in Table 1.
The transcription data was collected as described in Table 2. The transcription data set consists of 92 non-coding RNAs (denoted by gene IDs that begins with ’MAL’), while the rest are protein coding genes (denoted by gene IDs that start with ’PF3D7’). The feature to predict is DHA_IC50.
(Adapted from Turnbull et al., (2017) PLoS One11).
We used Apache Spark10 to pivot the dataset such that each isolate was its own row and each of the transcription values for each gene and attributes (i.e. timepoint, treatment, biological replicate) combination was its own column. This exercise transformed the training dataset from 272 rows and 5,546 columns to 30 rows and 44,343 columns, as shown in Table 3. We completed this pivot by slicing the data by each of the eight combinations of timepoint, treatment, and biological replicate, dynamically renaming the variables (genes) for each slice, and then joining all eight slices back together.
| Isolate | DHA_IC50 | hr24_trDHA_br1_Gene1 | hr24_trDHA_br2_Gene1 | … | hr6_trUT_br2_Gene5540 |
|---|---|---|---|---|---|
| isolate_01 | 2.177 | 0.008286 | -0.87203 | … | -2.24719 |
| … | … | … | … | … | … |
| isolate_30 | 1.363 | 0.195032 | 0.031504 | … | -1.72273 |
Example code shown below in the section labeled code 1. By using the massively parallel architecture of Spark, this transformation can be completed in a minimal amount of time on a relatively small cluster environment (e.g., <10 minutes using a 8-worker/36-core cluster with PySpark on Apache Spark 2.4.3).
1 ## Separate Dependent Variable 2 y =train.select(col("Isolate"), 3 col("DHA_IC50")) \ 4 .distinct() 5 6 ## Create Slice [Timepoint: 24HR, Treatment: DHA, BioRep: BRep1] 7 hr24_trDHA_br1 = train.drop("Sample_Name","DHA_IC50") \ 8 .filter((col("Timepoint") == "24HR") & 9 (col("Treatment") == "DHA") & 10 (col("BioRep") == "BRep1")) 11 ## Rename Columns 12 column_list = hr24_trDHA_br1.columns 13 prefix = "hr24_trDHA_br1_" 14 new_column_list = [prefix + s if s != "Isolate" else s for s in column_list] 15 16 column_mapping = [[o, n] for o, n in zip(column_list, new_column_list)] 17 18 hr24_trDHA_br1 = hr24_trDHA_br1.select(list(map(lambda old, new: col(old) \ 19 .alias(new),*zip(*column_mapping)))) 20 21 ## Join Slices Together 22 data = y.join(hr24_trDHA_br1, "Isolate", how='left') \ 23 .join(hr24_trDHA_br2, "Isolate", how='left') \ 24 .join(hr24_trUT_br1, "Isolate", how='left') \ 25 .join(hr24_trUT_br2, "Isolate", how='left') \ 26 .join(hr6_trDHA_br1, "Isolate", how='left') \ 27 .join(hr6_trDHA_br2, "Isolate", how='left') \ 28 .join(hr6_trUT_br1, "Isolate", how='left') \ 29 .join(hr6_trUT_br2, "Isolate", how='left')
Lastly, the dataset is then vectorized using the Spark VectorAssembler, and converted into a Numpy12-compatible array. Example code shown below in Code 1. Vectorization allows for highly scalable parallelization of the machine learning modeling in the next step.
1 ## Transform Data using VectorAssembler 2 assemblerInputs = numericalColumns 3 assembler = VectorAssembler(inputCols = assemblerInputs, outputCol="features") \ 4 .setHandleInvalid("keep") 5 stages += [assembler] 6 7 prepPipeline = Pipeline().setStages(stages) 8 pipelineModel = prepPipeline.fit(data) 9 vectordata = pipelineModel.transform(data) \ 10 .select(col("DHA_IC50"), col("features")) \ 11 .withColumnRenamed("DHA_IC50","label") 12 13 ## Convert to Numpy Array 14 import numpy as np 15 pddata = vectordata.toPandas() 16 seriesdata = pddata['features'].apply(lambda x : np.array(x.toArray())) \ 17 .as_matrix().reshape(-1,1) 18 X_train = np.apply_along_axis(lambda x : x[0], 1, seriesdata) 19 y_train = pddata['label'].values.reshape(-1,1).ravel() 20 21 ## Example Output (X_train) After Vectorization 22 array([[-0.62161893, -0.60860881, -1.11331369, ..., -1.457377 , 23 -3.292903 , -1.869169 ], 24 [-0.55719008, -2.41660489, -1.39244109, ..., -1.770098 , 25 -3.698841 , -1.740082 ], 26 ..., 27 [-0.17072536, -2.32828532, -1.08406554, ..., -1.402658 , 28 -5.314896 , -1.328886 ], 29 [-0.1923732 , -1.88763881, -1.23867258, ..., -1.971246 , 30 -3.567355 , -1.904116 ]])
We used the Microsoft Azure Machine Learning Service13 as the tracking platform for retaining model performance metrics as the various models were generated. For this use case, 498 machine learning models were trained using various scaling techniques and algorithms. We then created two ensemble models of the individual models using Stack Ensemble and Voting ensemble methods. Scaling and normalization methods are shown in Table 14.
The Microsoft AutoML package14 allows for the parallel creation and testing of various models, fitting based on a primary metric. For this use case, models were trained using Decision Tree, Elastic Net, Extreme Random Tree, Gradient Boosting, Lasso Lars, LightGBM, RandomForest, and Stochastic Gradient Decent algorithms along with various scaling methods from Maximum Absolute Scaler, Min/Max Scaler, Principal Component Analysis, Robust Scaler, Sparse Normalizer, Standard Scale Wrapper, Truncated Singular Value Decomposition Wrapper (as defined in Table 14). All of the machine learning algorithms are from the scikit-learn package15 except for LightGBM, which is from the LightGBM package16. The settings for the model sweep are defined in Table 4. The ‘Preprocess Data?’ parameter enables the scaling and imputation of the features in the data.
Once the 498 individual models were trained, two ensemble models (voting ensemble and stack ensemble) were then created and tested. The voting ensemble method makes a prediction based on the weighted average of the previous models’ predicted regression outputs whereas the stacking ensemble method combines the previous models and trains a meta-model using the elastic net algorithm based on the output from the previous models. The model selection method used was the Caruana ensemble selection algorithm17.
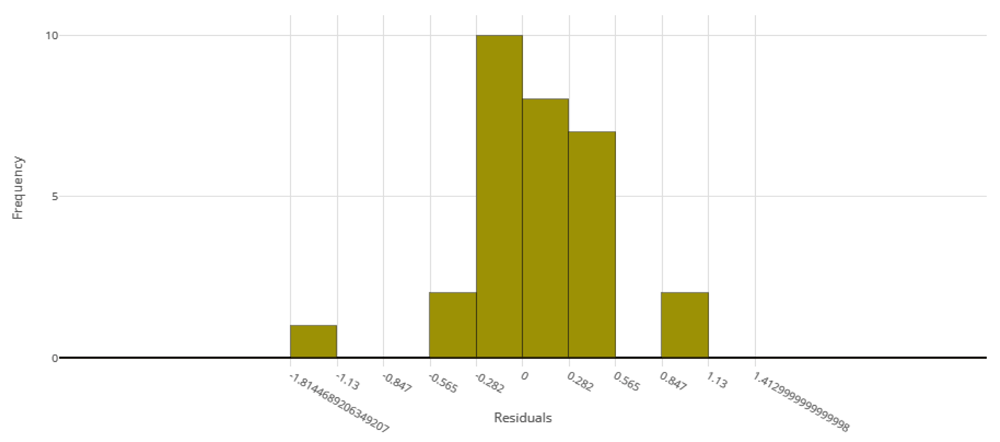
The voting ensemble model (using soft voting) was selected as the best model, having the lowest normalized Root Mean Squared Error (RMSE), as shown in Table 5. The top 10 models trained are reported in Table 6. Having a normalized RMSE of only 0.1228 and a Mean Absolute Percentage Error (MAPE) of 24.27%, this model is expected to accurately predict IC50 in malaria isolates. See Figure 1 for a visualization of the experiment runs and Figure 2 for the distribution of residuals on the best model.
Note that the top performing model (VotingEnsemble) is the final IC50 model discussed in this paper.
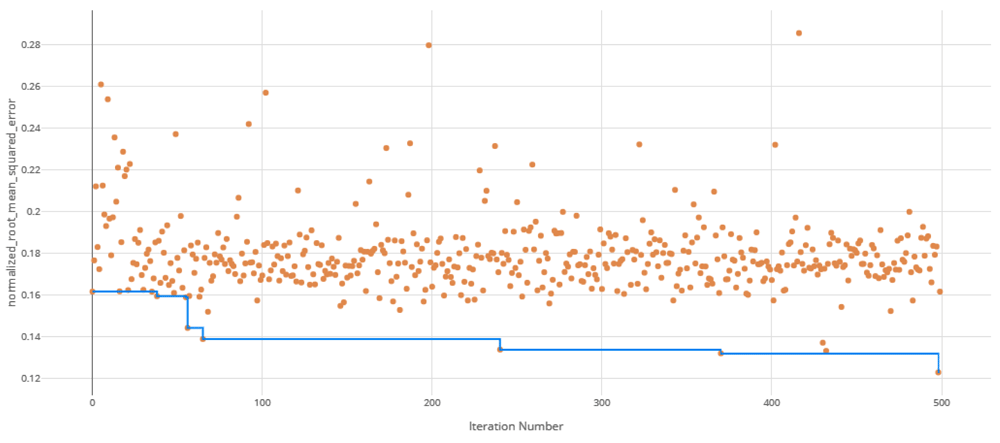
Each orange dot is an iteration with the blue line representing the minimum RMSE up to that iteration.
The second task of this work was to create a machine learning model that can predict the parasite clearance rate (fast versus slow) of malaria isolates. When resistance rates change in a pathogen, it can be indicative of regulatory changes in the pathogen’s genome. These changes can be exploited for the prevention of further resistance spread. Thus, a goal of this work is to understand genes important in the prediction of artemisinin resistance.
An in vivo transcription data set from Mok et al., (2015) Science18 was used to predict the parasite clearance rate of malaria parasite isolates based on in vitro transcriptional profiles (see Table 8).
The training data consists of 1,043 isolates with 4,952 genes from the malaria parasite Plasmodium falciparum. For each malaria parasite isolate, transcription data was collected for various PF3D7 genes. The form of the training dataset contains 1,043 rows and 4,957 columns, as shown in Table 7. The feature to predict is ClearanceRate.
The training data for this use case did not require the same pivoting transformations as in the last use case as each record describes a single isolate. Thus, only the vectorization of the data was necessary, which was performed using the Spark VectorAssembler and then converted into a Numpy-compatible array12. Example code is shown below. Note that this vectorization only kept the numerical columns, which excludes the Country, Kmeans_Grp, and Asexual_stage__hpi_ attributes as they are either absent or contain non-matching factors (i.e. different set of countries) in the testing data.
1 ## Transform Data using VectorAssembler 2 assemblerInputs = numericalColumns 3 assembler = VectorAssembler(inputCols = assemblerInputs, outputCol="features") \ 4 .setHandleInvalid("keep") 5 stages += [assembler] 6 7 prepPipeline = Pipeline().setStages(stages) 8 pipelineModel = prepPipeline.fit(data) 9 vectordata = pipelineModel.transform(data) 10 11 ## Convert to Numpy Array 12 import numpy as np 13 pddata = vectordata.toPandas() 14 seriesdata = pddata['features'].apply(lambda x : np.array(x.toArray())) \ 15 .as_matrix().reshape(-1,1) 16 X_train = np.apply_along_axis(lambda x : x[0], 1, seriesdata) 17 y_train = pddata['label'].values.reshape(-1,1).ravel() 18 19 ## Example Output (X_train) After Vectorization 20 array([[ 0.2263112 , -0.39682897, -1.80458125, ..., nan, 21 -1.30952803, -0.64170958], 22 [ 0.55442743, 0.54200115, -1.56157279, ..., 1.83083869, 23 0.21021662, -1.06553341], 24 ..., 25 [ 1.24446867, -0.09076431, -1.62156926, ..., 3.18060844, 26 -0.43056353, nan], 27 [ 0.99909549, -1.47208829, -1.91898139, ..., 2.59463935, 28 -1.21233458, nan]])
Once the 98 individual models were trained, two ensemble models (voting ensemble and stack ensemble) were then created and tested as before. Model search parameters are shown in Table 9.
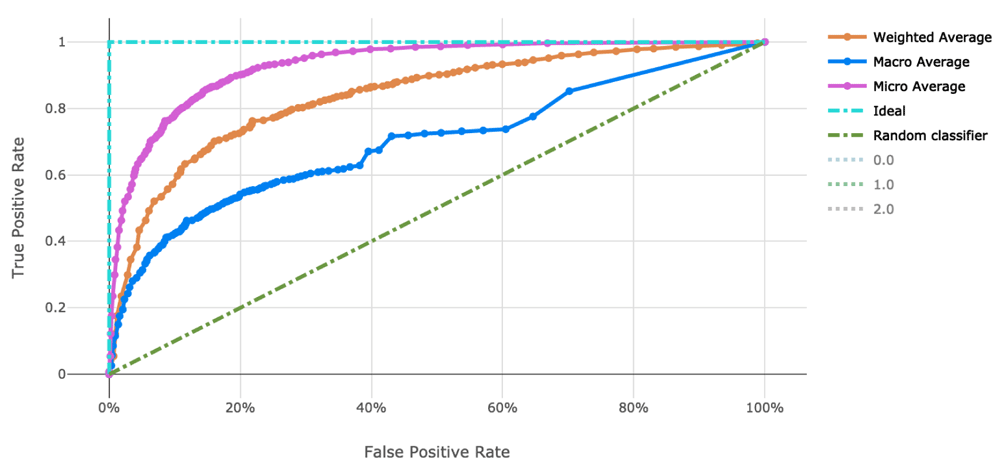
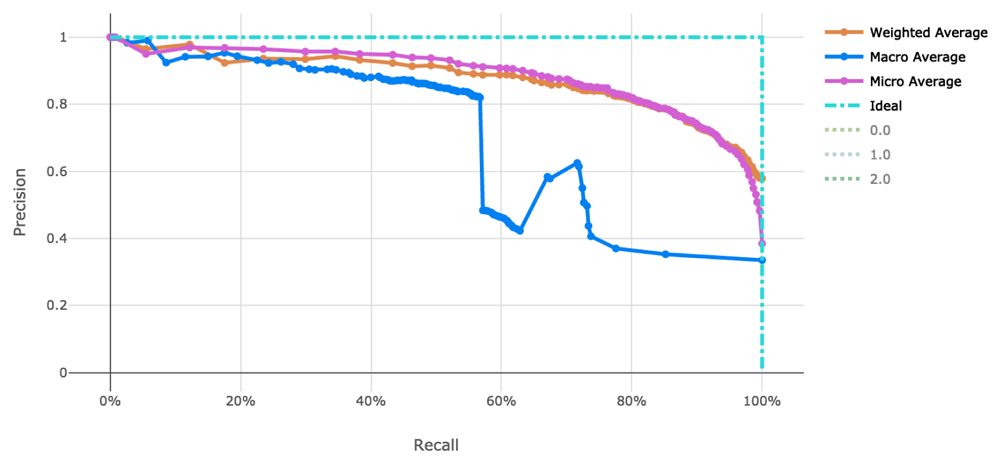
The voting ensemble model (using soft voting) was selected as the best model, having the highest area under the receiver operating characteristic curve (AUC), as shown in Table 11. The top 10 of the 100 models trained are reported in Table 10. Having a weighted AUC of 0.87 and a weighted F1 score of 0.80, this model is expected to accurately predict isolate clearance rates. A confusion matrix of the predicted results versus actuals is shown in Table 12. See Figure 3 for a visualization of the experiment runs and see Figure 4 and Figure 5 for the ROC and Precision-Recall curves on the best model.
Note that the top performing model (VotingEnsemble) is the clearance rate model discussed in this paper.
| Class | Prediction | |||
|---|---|---|---|---|
| Fast (ID: 0) | Slow (ID: 1) | Null (ID: 2) | ||
| Actual | Fast (ID: 0) | 661 | 74 | 0 |
| Slow (ID: 1) | 115 | 184 | 0 | |
| Null (ID: 2) | 6 | 3 | 0 | |

Each orange dot is an iteration with the blue line representing the maximum AUC up to that iteration.
subsectionFeature importance Feature importances were calculated using mimic-based model explanation of the ensemble model19. The mimic explainer works by training global surrogate models to mimic blackbox models. The surrogate model is an interpretable model, trained to approximate the predictions of a black box model as accurately as possible. See Figure 6 and Table 13.
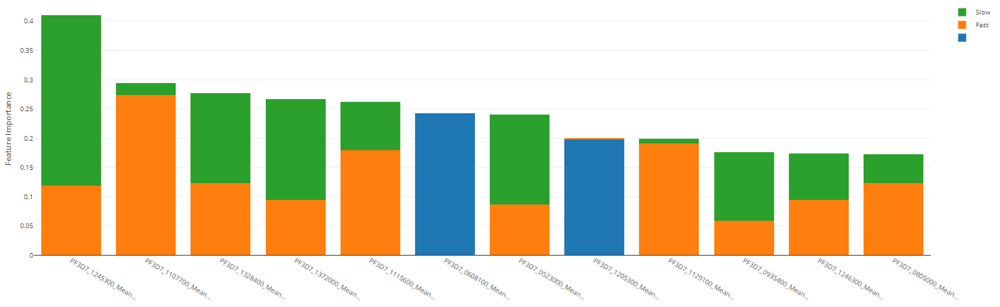
By using distributed processing of the data preparation, we can successfully shape and manage large malaria datasets. We efficiently transformed a matrix of over 40,000 genetic attributes for the IC50 use case and over 4,000 genetic attributes for the resistance rate use case. This was completed with scalable vectorization of the training data, which allowed for many machine learning models to be generated. By tracking the individual performance results of each machine learning model, we can determine which model is most useful. In addition, ensemble modeling of the various singular models proved effective for both tasks in this work.
The resulting model performance of both the IC50 model and the clearance rate model show relatively adequate fitting of the data for their respective predictions. While additional model tuning may provide a lift in model performance, we have demonstrated the utility of ensemble modeling in these predictive use cases in malaria.
In addition, this exercise helps to quantify the importance of genetic features, spotlighting potential genes that are significant in artemisinin resistance. The utility of these models will help in directing development of alternative treatment or coordination of combination therapies in resistant infections.
An earlier version of this article can be found on bioRxiv (doi:10.1101/856922).
The challenge datasets are available from Synapse (https://www.synapse.org/; Synapse ID: syn18089524). Access to the data requires registration and agreement to the conditions for use at: https://www.synapse.org/#!Synapse: syn18089524.
Challenge documentation, including the detailed description of the Challenge design, data description, and overall results can be found at: https://www.synapse.org/#!Synapse:syn16924919/wiki/583955.
Whole genome expression profiling of artemsinin-resistant Plasmodium falciparum field isolates, Accession number GSE59099: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE59099.
Zenodo: colbyford/malaria_DREAM2019: Ensemble Machine Learning Modeling for the Prediction of Artemisinin Resistance in Malaria - Initial Code Release for Research Publication (F1000). https://doi.org/10.5281/zenodo.359045921.
This project contains the following underlying data:
/SubChallenge1/data/sc1_X_train.pkl (Pickle file of the SubChallenge 1 independent variables, pivoted by Timepoint, Treatment, and BioRep.)
/SubChallenge1/data/sc1_y_train.pkl (Pickle file of the SubChallenge 1 dependent variable, DHA_IC50.)
/SubChallenge2/data/sc2_X_train.pkl (Pickle file of the SubChallenge 2 independent variables.)
/SubChallenge2/data/sc2_y_train.pkl (Pickle file of the SubChallenge 2 dependent variable, ClearanceRate.)
Data are available under the terms of the Creative Commons Zero "No rights reserved" data waiver (CC0 1.0 Public domain dedication).
Source code available from: https://github.com/colbyford/malaria_DREAM2019
Archived source code at time of publication: https://doi.org/10.5281/zenodo.359045921
License: GPL-3.0
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)