Keywords
epigenetics, CpG methylation, colon cancer, decitabine
This article is included in the Oncology gateway.
epigenetics, CpG methylation, colon cancer, decitabine
Colorectal cancer (CRC) is the third most common type of cancer worldwide, yet it is often caught only in its late stages1. While CRC incidence has been decreasing for individuals above 50 years of age, the incidence rates increased by 22% between 2000–2013 in individuals below 502. Chemotherapy is not effective, and patients typically survive only 44 months after the completion of treatment3. Despite this pressing reality, there is still debate about the molecular mechanisms underlying this disease. Genetic and epigenetic factors contribute to oncogenesis, but there is no consensus on a definitive molecular pathway4.
Cytosine methylation changes the expression of genes involved in cancer progression. DNA methylation is the transfer of a methyl group onto the C5 position of cytosine to form 5-methylcytosine5. Promoter hypermethylation has been observed to drive CRC disease progression. For instance, promoter hypermethylation of the hMLH1 gene leads to mismatch repair defects and a hypermutator phenotype in CRC6. Genetic knockout of two DNA methyltransferase genes restores expression of the tumor suppressor gene CDKN2A and slows growth of the CRC cell line HCT1167.
Numerous genomic regions have been shown to be consistently hypermethylated in multiple samples of colon cancer in comparison to regular colon cells8,9. For instance, Simmer et al.10, found 2867 genomic regions consistently differentially methylated regions in CRC (DMRs). However, the genome-wide methylation techniques used by these studies lack nucleotide-level resolution at these DMRs11, and do not address if these DMRs play a mechanistic role in CRC progression.
We sought to investigate if the colorectal cancer cell line, HCT-116, can serve as a viable model for DMR hypermethylation. HCT-116 cells have been observed to be an orthotopic model for colon cancer in mice12. Furthermore, knockout of the DNA methyltransferases DNMT1 and DNMT3b has been observed to deplete >98% of total methylation and slow HCT-116 growth7, indicating global DNA methylation plays a role in HCT-116 cell survival. If HCT-116 cells are to serve as a model for DMR hypermethylation in CRC, then we expect to observe hypermethylation at established high-confidence DMRs and a reversibility of methylation with the demethylating anticancer drug, decitabine13.
In the present study, we sought to investigate the effect of the decitabine on high confidence DMRs found in the promoter regions of genes previously connected to cancer progression from 2648 available DMRs. We selected DMRs proximal to DKK3, EN1, mir34b, SDC2, SPG20, and TLX1. DKK3 is a Wnt signaling pathway inhibitor found to be hypermethylated in colon cancer cells, possibly promoting oncogenic Wnt signaling14. EN1 is a canonical gene in development15 but has recently been linked to increased cell proliferation as a non-canonical prosurvival transcription factor in cancer cells16. In addition, the EN1 promoter is found to be hypermethylated in CRC with a CpG island methylator phenotype5. mir34b is a micro-RNA that is essential for normal brain development, motile ciliogenesis and spermatogenesis17. It was found to be hypermethylated in 100 out of 101 colon cancer cell lines18. SDC2 is a transmembrane proteoglycan that mediates cytoskeletal organization and adhesion to the ECM19. SDC2 acts as a positive regulator of growth factor signals whose aberrant expression correlates with tumor size20. SDC2 promoter is hypermethylated in cancer21. SPG20 regulates cytokinesis and may alter cell division when aberrantly methylated in CRC22. TLX1 functions as a transcription factor23 and its gene promoter is frequently hypermethylated in CRC8,10. While it is overexpressed and demethylated in leukemia24 aberrant hypermethylation may also promote growth, as reviewed in 25.
We predicted that decitabine will have a negative effect on HCT-116 proliferation, and will decrease hypermethylation at these DMRs. Using the UCSC genome browser, we observed that DMR methylation inversely correlated with transcriptional activator binding across multiple cell lines. Using bisulfite PCR we observed that decitabine inhibits methylation across each DMR.
DMRs were selected from Simmer et al.10. Genomic regions from table S4 of that publication were aligned to human genome 19 (hg19) using liftOver26. UCSC genome browser was configured to visualize transcription factors and methylation status according to the following configuration: http://genome.ucsc.edu/s/williamhconrad/hg19%2Dall%2Dcell%2Dmethylation. Transcription factors were selected randomly from the track UCSC genome browser track "Transcription Factor ChIP-seq (161 factors) from ENCODE with Factorbook Motifs". 10 cell lines with transcription factor binding were recorded and 10 cell lines lacking transcription factor binding were recorded at each DMR. Cell lines were selected using a random number generator. Each transcription factor was recorded as a transcriptional repressor or enhancer according to UniProt27. The methylation status was then recorded for each of those cell lines. Methylation was recorded as fully methylated, mostly methylated, majority methylated, half-methylated, minority methylated, mostly unmethylated, fully unmethylated, not detected, or not tested. The cell lines were then sorted into methylated or unmethylated. The number of repressors and enhancers was recorded, and a chi squared analysis was performed to compare differences in repressor and enhancer binding to methylated or unmethylated regions using excel Data are published as underlying data28.
HCT-116 cells were obtained from American type culture collection (ATCC; CCL-247) and cultured in McCoy’s 5A media (ATCC; 30-2007) in 10% fetal bovine serum (ATCC; 30-2007) and penicillin streptomycin (Thermo; 15140122). Cells were maintained in tissue culture flasks at 37 °C and 5% CO2 according to established protocols from ATCC29.
To test the effect of decitabine on the clonogenic survival of HCT-116 cells, we used protocols adapted from Franken et al.30 and Palii et al.31. Cells were plated at a density of 200 per well in a 6-well tissue culture plate. After overnight incubation, cells were treated with vehicle (anhydrous DMSO; Sigma; 276855), 0.1, 0.25, or 1 µM decitabine (Sigma; A3656). Cells were treated again after 24 hrs and then replaced with 2 ml of complete McCoy’s 5A media. After 12 days of growth, cells were fixed and stained with 10% w/v glutaraldehyde (Sigma; 340855), 0.5% w/v crystal violet (Sigma; C6158) in PBS (Sigma; 1408). After extensive rinsing in tap water, colonies were counted by eye. The average number of colonies was compared across the four conditions using a one-way ANOVA followed by Tukey’s HSD post hoc test.
HCT-116 cells were cultured as described above. Exponentially growing cells were passaged to a density 20% confluency. The day following passage, cells were treated with 1 µM decitabine or DMSO. After 24 hours, treatment was repeated. After four additional days of incubation, cells were collected by trypsinization. 400,000 cells for each condition were collected and genomic DNA was isolated using the PureLink genomic DNA mini kit according to the manufacturers instructions (Thermo Fisher; K182001).
400 ng of genomic DNA isolated as above was treated with 10 units of HpaII (NEB; R0171S) at 37 C for 1 hr. genomic DNA digestion was then evaluated by 1% agarose (VWR; 97062-244) gel electrophoresis. DNA was stained with 1x sybr safe (VWR; 470193-138) and imaged on a Bio-rad chemidoc imaging system.
400 ng of genomic DNA isolated as above was bisulfite converted using the EZ DNA methylation kit according to the manufacturer’s instructions (zymo research; D5001). Bisulfite converted DNA was eluted in 10 µl at a concentration of approximately 40 ng / µl. Oligonucleotides for bisulfite PCR were designed using MethPrimer32. The positive strand sequence for a DMR was collected from UCSC genome browser and primers were designed for an amplicon between 150 and 400 nucleotides. Optimal annealing temperature for each primer pair were determined by testing a range of annealing temperatures between 44 and 66 °C for each primer pair against fully unmethylated and fully methylated genomic DNA (zymo; D5014). Primer pairs (Table 1), optimal annealing temperatures (Table 1), and PCR reaction conditions (Table 2), reaction master mix (Table 3) are provided in the indicated tables. Amplified DNA was purified from oligonucleotide primers and dNTPs using zymo DNA clean and concentrator-5 according to the manufacturer’s instructions (zymo; D4013). Samples were eluted in 10 µl of elution buffer and submitted for sequencing at the University of Chicago Comprehensive Cancer Center DNA sequencing and genotyping facility (Chicago, IL). Percent methylation was calculated using the relative peak height of cytosine and uracil at a given CpG site, as described previously. Peak height was quantified using Thermo Fisher Variant analysis app on the thermo fisher connect web site. Briefly, this cloud-based application processes .abi sequencing chromatogram files and returns base calls and peak height values for each peak on the chromatogram. The open source software Chromaseq can also extract identical base call and peak height values from .abi files33. Briefly, chromaseq can be installed according to their web site. The .abi sequencing files can be viewed using this software. Selecting a base call will reveal the identical peak height value presented in the chromatogram as exported in the Thermo Fisher Variant analysis app.
| step | cycle | temp (C) | time (m:ss) |
|---|---|---|---|
| 1 | denaturing | 95 | 5:00 |
| 2 | denaturing | 95 | 0:30 |
| 3 | annealing | See Table 1: | 0:45 |
| 4 | extension | 68 | 0:45 |
| 5 | go to step 2 39 times | ||
All statistical analyses were performed using GraphPad Prism version 7.0d. To evaluate the effect of various doses of decitabine on clonogenic survival against a no-decitabine control, a one-way ANOVA with Tukey’s post hoc was performed to control for multiple comparisons (i.e. all drug conditions against the same control). To evaluate the percent methylation of DMRs in the presence or absence of decitabine, a one-way ANOVA was performed with Bonferroni’s post hoc test to allow for multiple comparisons (i.e. between control and decitabine treated for each DMR). The specific statistical tests used are also described in the figure legends.
In mammals, cytosine methylation inversely correlates with gene expression34,35. We sought to determine if our selected DMRs might regulate gene expression. Using the UCSC genome browser36, we identified transcription factor binding data in our selected DMR regions from the encyclopedia of DNA elements (ENCODE) project37. The ENCODE project has performed 2041 transcription factor CHIP-seq experiments across 90 cell lines38,39. Transcription factor binding was observed at our DMRs (Figure 1). We hypothesized that if methylation silenced gene expression at the DMRs we selected, then we would see diminished binding of transcriptional enhancers in cell lines with methylation present at that DMR. For each of our six DMRs we observed TF binding across 20 cell lines selected by random number generator (see methods). We categorized the degree of TF binding and degree of methylation for these 10 cell lines (Underlying data for Table 428).
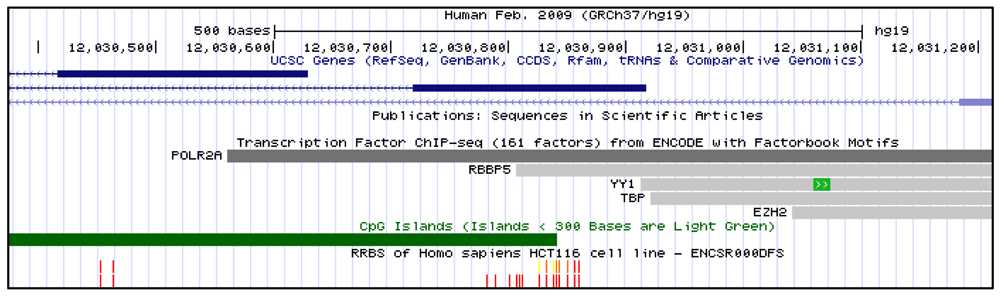
Representative screen capture of the UCSC genome browser view of the DKK3 DMR. Scale depicted in track 1 (black). DKK3 gene depicted in track 2 (blue). Transcription factor binding observed in ENCODE database depicted in track 3 (grey). CpG islands depicted in track 4 (green). CpG methylation as observed by reduced representation bisulfite sequencing in HCT-116 cells depicted in track 5 (red is 100% methylated, green is 0% methylated).
Indeed, we observed no transcriptional enhancers bound to methylated DMRs. Interestingly, binding of transcriptional repressors was also diminished at methylated DMRs, perhaps because methylation supplants the need for transcription factor repression. In general, transcriptional repressors and enhancers both bound more readily to cell lines with unmethylated DMRs (Table 4). The differences between repressor and enhancer binding in methylated and unmethylated DMRs was significantly different by chi-squared analysis (p<0.05). From these data, we conclude that methylation of the selected DMRs repress transcriptional enhancer binding across a broad range of cell lines.
At each DMR, transcription factor binding was evaluated across cell lines tested in the ENCODE project. No transcriptional activators were detected at methylated DMRs across cell lines. Fewer repressors were also detected at methylated DMRs. However, repressors failed to bind methylated and unmethylated DMRs with similar frequency.
| Repressor binds | Enhancer binds | |||
|---|---|---|---|---|
| Y | N | Y | N | |
| methylated | 1 | 8 | 0 | 7 |
| unmethylated | 21 | 8 | 33 | 22 |
The ability of a cancer cell to form a colony has been a long-standing measure for its survival in the host. Interventions that ablate clonogenicity increase patient survival40. Decitabine is known to inhibit clonogenic survival of HCT-116 cells31, and we observe inhibition of clonogenic survival at similar doses ranging from 1 µM to 100 nM (Figure 2). Furthermore, decitabine treatment is known to increase sensitivity of HCT-116 genomic DNA to the restriction enzyme, HpaII, which is inhibited by CpG methylation7. Likewise, we observe that genomic DNA collected from HCT-116 cells treated for 48 hr with 1 µM decitabine was digested by decitabine (Figure 3). From these data, we conclude that decitabine inhibits the clonogenic survival of HCT-116 cells and also inhibits DNA methylation.

Number of HCT-116 colonies detected by crystal violet staining 14 days after two 24-hour treatments of the indicated doses of decitabine. *p<0.05 ANOVA, Tukey's post-hoc.

HCT-116 cells were treated with 1 µM decitabine or DMSO for 48 hrs. Genomic DNA was extracted and treated with the methylation-sensitive enzyme, HpaII as indicated. 400 ng of gDNA was separated by gel electrophoresis. Ladder indicated to the left of the gel.
After observing global demethylation by 1 µM decitabine (Figure 3), we next sought to determine the degree of CpG methylation at DMRs in HCT-116 cells, and if those methylation sites were inhibited by decitabine. Using bisulfite PCR, we detected conversion of unmethylated cytosine to uracil (Figure 4a). Importantly, we both identified methylated CpG sites previously detected by reduced-representation bisulfite sequencing as part of the ENCODE project, and we also identified novel CpG sites in the region, adding resolution to the methylation status of these select DMRs (Figure 4b).
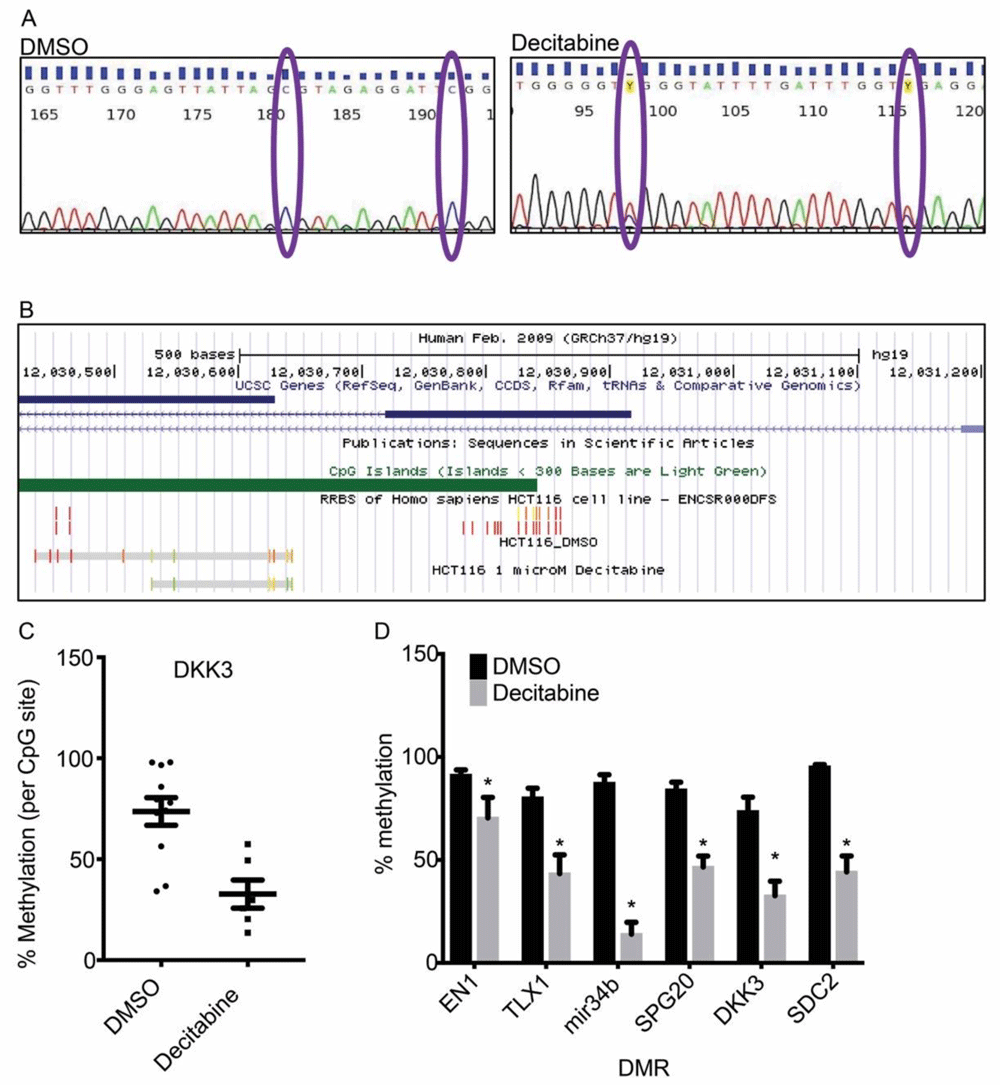
(a) Representative chromatograms of bisulfite PCR / sanger sequencing of the DKK3 DMR. Percent methylation quantified by peak height of C relative to C + T. (b) Mapping of bisulfite PCR results onto human genome 19. Scale depicted in track 1 (black). DKK3 gene depicted in track 2 (blue). CpG islands depicted in track 3 (green). CpG methylation as observed by reduced representation bisulfite sequencing in HCT-116 cells depicted in track 4 (red is 100% methylated, green is 0% methylated). Bisulfite PCR depicted in track 5 (DMSO) and track 6 (decitabine treated). The two grey bars show region sequenced. The red-green spectrum depict degree of methylation. (c) Quantification of percent methylation of sites depicted in (b). Percent methylation for each DMR as in (c). *p<0.05 ANOVA with Bonferroni’s post-hoc test. Columns and bars are mean and SEM, respectively.
We quantified the degree of CpG methylation at each site detected by bisulfite PCR in the presence or absence of decitabine. We observed statistically significant reductions in CpG methylation at all tested DMRs. From these data we can conclude that all colon cancer DMRs tested are hypermethylated in HCT-116 cells, that bisulfite PCR offers increased resolution at DMRs over HM450 array or RRBS, and that methylation at DMRs is reversable by 1 µM decitabine treatment.
From our results, we conclude that HCT-116 cells can serve as a model for investigating the role of high confidence DMRs in colon cancer. As observed previously, we also observed HCT-116 cells to be sensitive to the demethylating agent decitabine7,31. Decitabine inhibited clonogenicity (Figure 2) and demethylated genomic DNA (Figure 3). By bisulfite PCR, we observed DMRs previously identified in patient colon cancer cells to be consistently hypermethylated in HCT-116 cells. Furthermore, we achieved single-nucleotide resolution of CpG methylation at these DMRs. We were able to confirm previously identified CpG sites as well as identify new ones (Figure 4).
The HCT-116 model of DMR methylation has potential to shed light on the role of DMRs in colon cancer pathogenesis. Moving forward, it will be important to determine if (a) DMRs reduce nearby gene expression, (b) if DMR demethylation increases gene expression, and (c) if restoring expression of methylation-silenced genes affects HCT-116 growth or survival. In future, we plan to increase resolution at DMRs relevant to HCT-116 cell growth by sequencing across the entire DMR and by testing additional DMRs. The most updated data can be viewed using the UCSC genome browser public session found here: https://bit.ly/UCSC-DMR-methylation. The original data presented in this manuscript will be maintained as described in the data availability section28,41.
Future work is needed to address the question if specific gene products suppressed by hypermethylation play a role in tumor growth. Nine frequently hypermethylated genes have been observed to slow cancer cell growth when heterologously expressed42. Two additional frequently hypermethylated genes have been observed to inhibit cancer cell colony formation when heterologously expressed43. Moving forward, we seek to heterologously express gene products suppressed by hypermethylation to test if such expression affects tumor cell growth and colony formation.
Zenodo: williamhconrad/HCT116-DMR-bisulfite-PCR: bisulfite PCR repository under CC0 license. http://doi.org/10.5281/zenodo.394843928
This project contains the following underlying data:
“Underlying data for Figure 2 - clonogenic survival.xlsx” (a spreadsheet containing the clonogenic survival data depicted in figure 2)
“Underlying data for Figure 4b - UCSC methylation tracks for DMSO.bed” (a spreadsheet (tab-delimited bed format) containing the percent methylation data for DMSO treated cells presented in figure 4b)
“Underlying data for Figure 4b - UCSC methylation tracks for decitabine.bed” (a spreadsheet (tab-delimited bed format) containing the percent methylation data for decitabine treated cells presented in figure 4b)
“Underlying data for Figure 4c and d raw methylation quantification.xlsx” (a spreadsheet containing the raw methylation data data depicted in figures 4c and d)
“Underlying data for table 4 DMR TF dataset-FINAL.xlsx” (a spreadsheet containing the transcription factor binding data for DMRs evaluated in table 4)
fig 3 raw hpaii digest image 300 dpi.tif (Raw gel image for Figure 3)
Zenodo: williamhconrad/HCT116-decitabine-hub: Decitabine Hub repository under CC0 license. http://doi.org/10.5281/zenodo.394675341.
This project contains the following underlying data that are used to build the UCSC genome browser public hub found at https://bit.ly/UCSC-DMR-methylation:
“description.html” (An html file that describes the Tracks in this repository. This html file is used by UCSC genome browser to build a description for the public hub)
“description.fld” (A folder with formatting for the file “description.html”)
“genomes.txt” (A file used by UCSC genome browser to select the correct genome to annotate the methylation data)
“hub.txt” (A file used by UCSC genome browser to find the DNA methylation tracks )
“ BMB322L-pctMethyl-DMSO-20200602.bb”, “ BMB322L-pctMethyl-DMSO-20200602.bed”, and “ BMB322L-pctMethyl-DMSO-20200602.bed” (identical spreadsheets in three formats containing the percent methylation data for DMSO treated cells presented in figure 4b for use by UCSC genome browser. The “.bb” and “bigBed” files are in bigbed format, the “.bed” file is in bed format.)
“ BMB322L-pctMethyl-Decitabine-20200602.bb”, “ BMB322L-pctMethyl- Decitabine -20200602.bed”, and “ BMB322L-pctMethyl- Decitabine -20200602.bed” (identical spreadsheets in three formats containing the percent methylation data for Decitabine treated cells presented in figure 4b for use by UCSC genome browser. The “.bb” and “bigBed” files are in bigbed format, the “.bed” file is in bed format.)
Data are available under the terms of the Creative Commons Zero "No rights reserved" data waiver (CC0 1.0 Public domain dedication).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)